

A palavra câncer se aplica a mais de uma centena de doenças – algumas que evoluem rapidamente, outras que só se manifestam depois de décadas; algumas altamente curáveis, outras incontornavelmente fatais –, todas com uma característica em comum: a proliferação desenfreada das células. Com base em resultados internacionais e dados obtidos em seu laboratório na Universidade Federal do Rio de Janeiro (UFRJ), o bioquímico Jerson Lima da Silva formulou a hipótese de que ao menos parte dos casos de câncer seja desencadeada pelo mesmo mecanismo molecular que está por trás da doença de Creutzfeldt-Jakob, a versão humana do mal da vaca louca, que causa a morte celular precoce e deixa o cérebro poroso feito uma esponja.
De acordo com essa visão, defendida por Silva e seus colaboradores em um artigo publicado em junho na Bioscience Reports, tanto no câncer, marcado pela perpetuação da vida das células, como na doença de Creutzfeldt-Jakob, em que a morte celular é antecipada, a origem do problema seria a mesma: o enovelamento anormal de uma proteína. Pode parecer uma causa sutil demais para estragos tão grandes. Mas, acreditam os pesquisadores, faz sentido. Afinal, é a estrutura tridimensional dessas moléculas grandes e complexas, fundamentais para definir a estrutura e o funcionamento das células, que determina o papel que vão desempenhar. Quando o enovelamento dá errado, as proteínas em geral deixam de funcionar como deveriam e até ganham funções extras. A diferença entre os casos de câncer e os de Creutzfeldt-Jakob estaria na proteína afetada.
Nas formas de câncer analisadas pelo grupo da UFRJ, a deformação atinge a p53, proteína que já foi chamada de guardiã do genoma por coordenar a reparação do DNA no caso de pequenos danos e por encaminhar a célula para a morte quando os defeitos não podem ser consertados. Já na doença de Creutzfeldt-Jakob a proteína alterada é o príon celular, molécula que se ancora na superfície externa das células e controla o trânsito de informações do meio externo para o interno (ver Pesquisa FAPESP nº 148). Em ambos os casos, a falha no enovelamento parece conferir à proteína alterada uma característica típica de agentes infecciosos tradicionais como os vírus e as bactérias: a capacidade de se autopropagar e infectar outras células.
A ideia de que versões deformadas de uma proteína podem causar doenças não é nova. Foi proposta nos anos 1980 pelo pesquisador norte-americano Stanley Prusiner para explicar a origem do grupo de enfermidades neurodegenerativas do qual fazem parte a doença de Creutzfeldt-Jakob e o mal da vaca louca – as encefalopatias espongiformes. Investigando o agente causador de uma encefalopatia que atinge as ovelhas, Prusiner não encontrou os vírus que esperava. Em vez disso, identificou apenas uma proteína defeituosa à qual chamou de príon (sigla de partícula proteinácea infecciosa) e formulou uma explicação de como os príons deformariam as proteínas saudáveis. Segundo essa hipótese, que rendeu a Prusiner um prêmio Nobel em 1997, o simples contato da molécula deformada com as proteínas normais é suficiente para induzir uma transformação na estrutura tridimensional delas. É um evento em cadeia que, uma vez iniciado, não se consegue deter, como pedras de dominó que tombam. Também é um efeito difícil de reverter. As proteínas defeituosas têm uma estrutura mais estável do que as saudáveis e aderem umas às outras, originando longas fibras tóxicas para os neurônios.
“Acreditamos que o mesmo ocorra em uma parte dos casos de câncer em que a p53 está deformada”, contou Silva em seu laboratório no início de agosto, dias depois de um evento em que comentou seus resultados com Prusiner. Segundo Silva, estudos internacionais recentes sugerem ainda que algumas versões deformadas da p53 poderiam passar de uma célula a outra. Isso não significa, no entanto, que elas possam ser transmitidas entre indivíduos da mesma espécie. “Essas proteínas seriam transmissíveis [de célula para célula], mas não infecciosas”, explicou o bioquímico, que coordena o Instituto Nacional de Ciência e Tecnologia de Biologia Estrutural e Bioimagem e é o diretor científico da Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (Faperj).
Câncer de mama
As evidências mais contundentes de que versões defeituosas da p53 podem atuar como príon – os pesquisadores dizem que elas têm ação prionoide – e induzir a remodelagem das proteínas saudáveis, fazendo-as perder sua função original, surgiram nos últimos dois anos. Em parceria com a equipe da geneticista Cláudia Moura-Gallo, da Universidade do Estado do Rio de Janeiro, o grupo de Silva analisou amostras de tumores de mama de 88 mulheres e verificou que na maioria dos casos havia agregados formados por moléculas defeituosas da p53, semelhantes aos agregados amiloides das doenças causadas por príon – essas fibras ou agregados também aparecem em outras enfermidades neurodegenerativas, como a doença de Parkinson e a de Alzheimer. Quase sempre as proteínas deformadas haviam sido geradas em decorrência de pequenas alterações no gene TP53, que contém a receita para a produção dessa proteína.
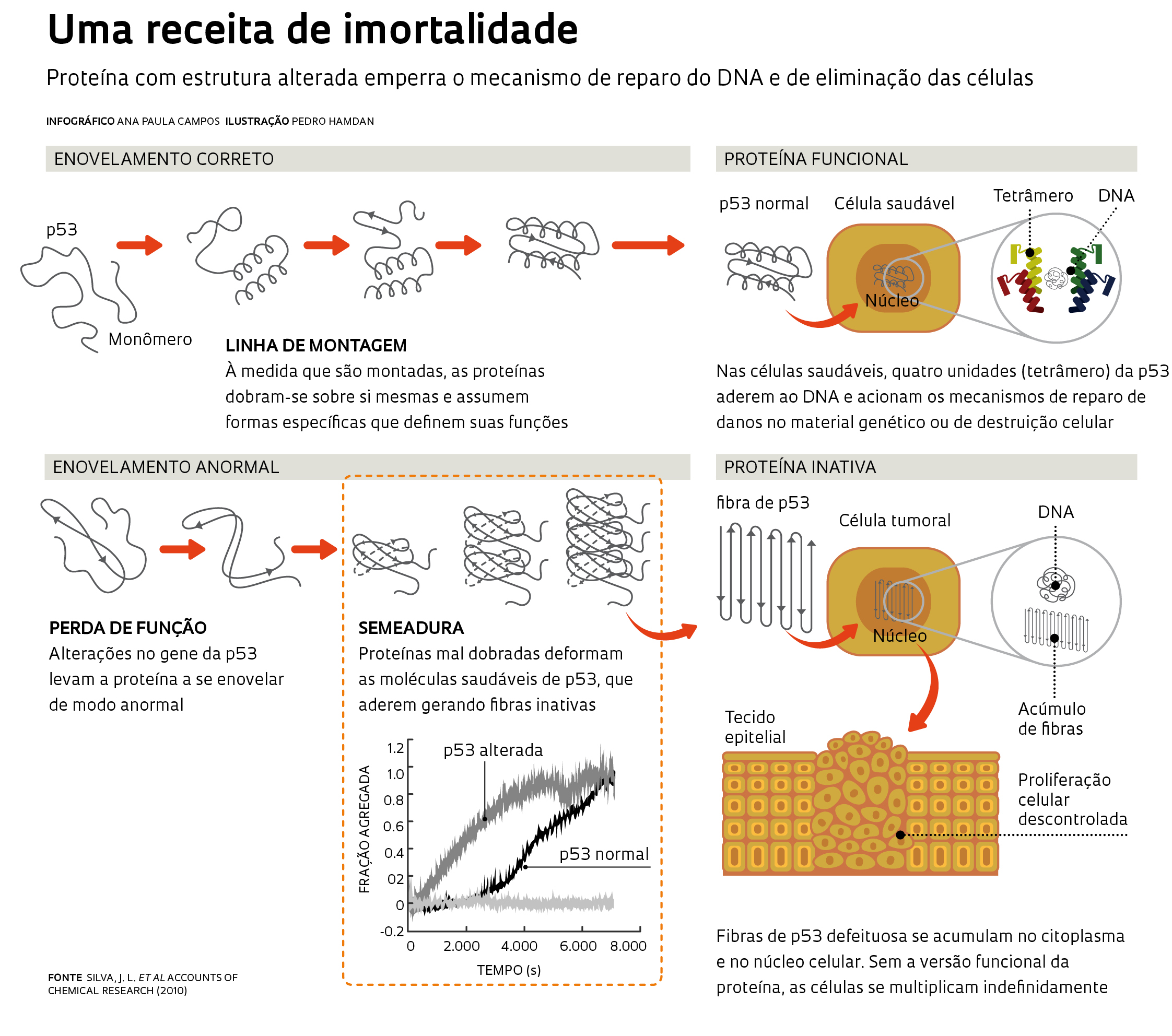 Era a primeira vez que se encontravam fibras de p53 em células tumorais. Mas a simples identificação dessas fibras não era suficiente para demonstrar que a proteína alterada podia disparar a deformação das proteínas saudáveis – fenômeno que em inglês recebe o nome de seeding ou semeadura, uma propriedade característica dos príons. No laboratório de Silva, a biomédica Ana Paula Ano Bom, a farmacêutica Luciana Rangel e a bioquímica Danielly Costa iniciaram, então, testes para identificar as condições em que a p53 – tanto sua versão saudável quanto a deformada – gerava os agregados. Para isso, mediram o tempo que leva para as fibras surgirem e a forma que assumem sob diferentes condições químicas (de pH) e físicas (de pressão e temperatura).
Era a primeira vez que se encontravam fibras de p53 em células tumorais. Mas a simples identificação dessas fibras não era suficiente para demonstrar que a proteína alterada podia disparar a deformação das proteínas saudáveis – fenômeno que em inglês recebe o nome de seeding ou semeadura, uma propriedade característica dos príons. No laboratório de Silva, a biomédica Ana Paula Ano Bom, a farmacêutica Luciana Rangel e a bioquímica Danielly Costa iniciaram, então, testes para identificar as condições em que a p53 – tanto sua versão saudável quanto a deformada – gerava os agregados. Para isso, mediram o tempo que leva para as fibras surgirem e a forma que assumem sob diferentes condições químicas (de pH) e físicas (de pressão e temperatura).
Elas constataram que as duas formas da p53 geravam espontaneamente os agregados em condições semelhantes às que as células encontram no corpo humano – temperatura de 37 graus Celsius e pH neutro ou um pouco ácido, típico dos tumores. A formação dessas fibras ocorria mesmo quando apenas o segmento central, a parte da p53 que interage com o DNA, era usado nos experimentos. A diferença é que o dos agregados tóxicos se formaram mais rapidamente a partir da p53 deformada, relataram os pesquisadores em agosto de 2012 no Journal of Biological Chemistry (ver infográfico acima).
O resultado mais importante surgiu quando as pesquisadoras adicionaram diminutas concentrações da proteína defeituosa em recipientes contendo p53 saudável. Como um sorvedouro, a proteína deformada atraiu as moléculas normais e induziu a sua conversão na forma alterada – confirmando sua propriedade de seeding. A capacidade de induzir a deformação foi maior quando se usou a versão da p53 alterada resultante de uma mutação conhecida como R248Q, uma das sete mais frequentes no câncer. “Encontramos agregados dessa forma mutante em amostras de câncer de mama e em linhagens de células desse tumor cultivadas em laboratório”, conta Danielly.
Pequenas alterações, grandes efeitos
A formação dos aglomerados de p53 parece não ser exclusiva dos tumores de mama. No início deste ano a equipe de Rakez Kayed, da Universidade do Texas, em Galveston, descreveu essas fibras em um tipo bastante frequente de tumor de pele, o carcinoma basocelular. Mais recentemente o grupo de Silva também as identificou em amostras de glioblastoma humano, um dos tumores cerebrais mais agressivos que se conhece.
 imagens ano bom, a.p. et al. journal of biological chemistry (2012)A tendência à agregação não ocorre só em consequência da mutação R248Q. Outras alterações pontuais no gene geram versões da proteína com propensão a se agregar, observaram os pesquisadores da UFRJ. Além de modificar a estrutura da p53 saudável, as versões deformadas dessa molécula sequestram e danificam duas outras proteínas da mesma família: a p63, que controla a multiplicação celular, e a p73, que, de forma independente, encaminha as células com defeito para a apoptose, um tipo de morte programada. “Essas duas proteínas também desempenham um importante papel antitumoral”, explica Danielly.
imagens ano bom, a.p. et al. journal of biological chemistry (2012)A tendência à agregação não ocorre só em consequência da mutação R248Q. Outras alterações pontuais no gene geram versões da proteína com propensão a se agregar, observaram os pesquisadores da UFRJ. Além de modificar a estrutura da p53 saudável, as versões deformadas dessa molécula sequestram e danificam duas outras proteínas da mesma família: a p63, que controla a multiplicação celular, e a p73, que, de forma independente, encaminha as células com defeito para a apoptose, um tipo de morte programada. “Essas duas proteínas também desempenham um importante papel antitumoral”, explica Danielly.
Em um estudo publicado em julho deste ano na PLoS One, Xavier Roucou e sua equipe na Universidade de Sherbrook, em Quebec, Canadá, demonstraram que uma versão defeituosa da p53 é, sim, capaz de infectar células saudáveis. Os pesquisadores acrescentaram moléculas deformadas a culturas de células e viram que as proteínas alteradas eram absorvidas por bolsas que se formavam na membrana. Uma vez no interior das células, a p53 mal dobrada desencadeou a formação de fibras.
Como toda ideia nova, a hipótese de que versões mutadas da p53 possam funcionar como príon não é consensual. “O trabalho da equipe da UFRJ é bastante consistente, mas são necessárias mais evidências”, comenta a bioquímica Vilma Martins, do Centro Internacional de Pesquisa do A.C. Camargo Cancer Center, em São Paulo, estudiosa das doenças causadas por príon. Um dos passos que falta para demonstrar a ação prionoide da p53 é avaliar se a proteína deformada em laboratório origina tumores em modelos animais. Apesar da cautela, Vilma acredita que esse mecanismo possa explicar a origem de parte dos casos de câncer ligados a mutações no gene TP53.
Se a ideia do grupo da UFRJ estiver correta, ela ajudará a entender o desenvolvimento dos tumores ditos espontâneos, que não são transmitidos de uma geração a outra e surgem em consequência de alterações genéticas no embrião já formado ou no indivíduo adulto. A ação prionoide da p53 seria uma boa explicação para os tumores espontâneos – a grande maioria dos casos de câncer –, em especial quando ocorre a chamada dominância negativa. “Nesse fenômeno biológico, a alteração de apenas uma das duas cópias de um gene já é suficiente para levar ao desenvolvimento de uma enfermidade”, explica Silva, que levantou a possibilidade no caso da p53 já em 2003, quando começou a estudar o enovelamento da proteína. Ele imagina ser possível usar as fibras de p53 em um teste, como marcador molecular de gravidade do câncer ou de prognóstico. E ainda que no futuro se torne possível interferir nesse mecanismo e tentar frear o desenvolvimento de alguns tumores. “Talvez”, diz, “se encontre uma forma de bloquear o processo de agregação”.
Artigos científicos
SILVA, J. L. et al. Expanding the prion concept to cancer biology: dominant-negative effect of aggregates of mutant p53 tumor suppressor. Bioscience Reports. 27 jun. 2013
SILVA, J. L. et al. Ligand binding and hydration in protein misfolding: insights from studies of prion and p53 tumor suppressor proteins. Accounts of Chemical Research. v. 43, n.2, p. 271-9. 2010.
ANO BOM, A. P. et al. Mutant p53 aggregates into prion-like, amyloid oligomers and fibrils: implications for cancer. Journal of Biological Chemistry. v. 287, n. 33, p. 28.152-62. 10 ago. 2012.