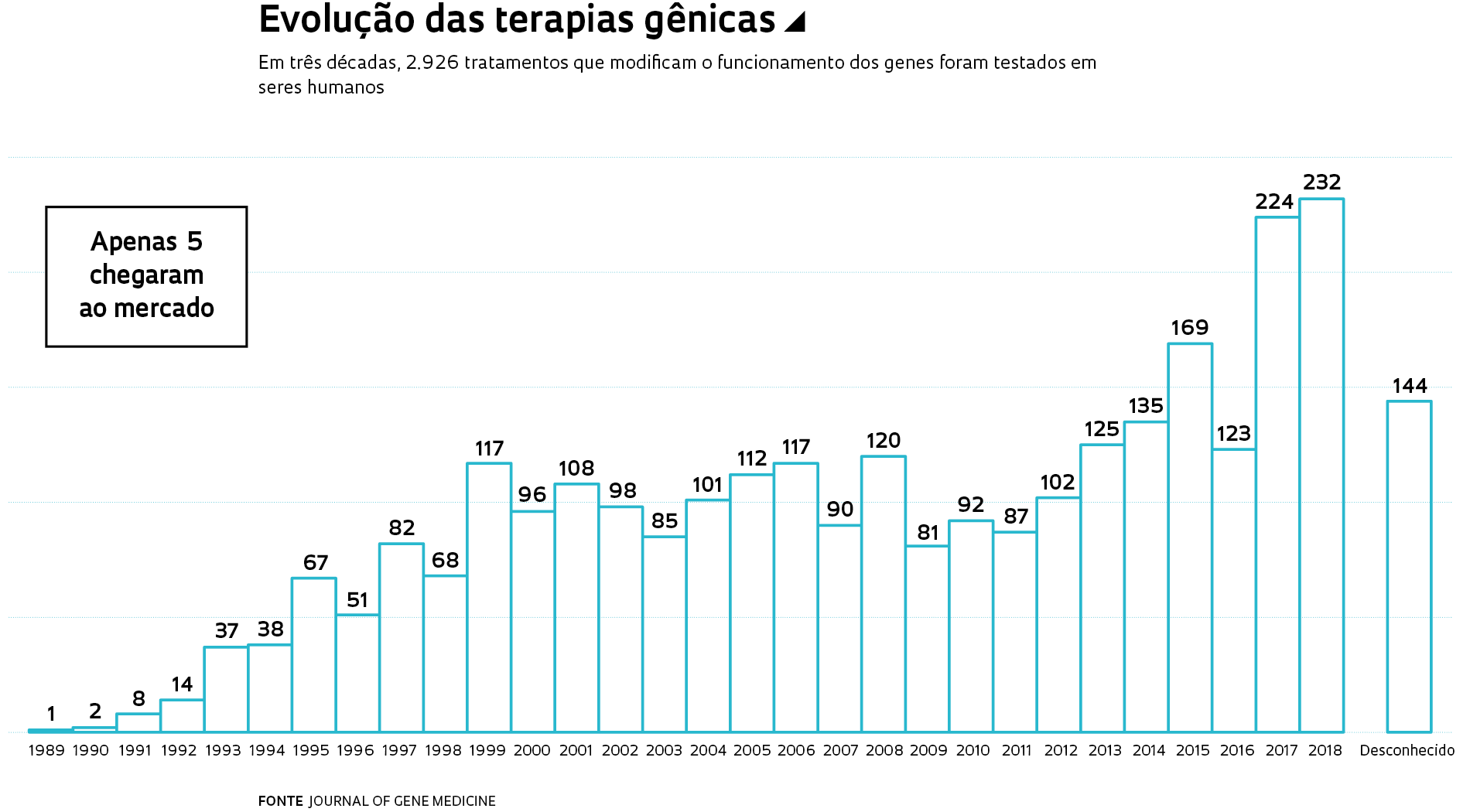
A partir deste mês, oito centros do sistema público de saúde brasileiro especializados em doenças raras devem disponibilizar para todas as crianças com atrofia muscular espinal (AME) o primeiro medicamento capaz de amenizar os sintomas do problema de origem genética. Esse tipo de atrofia leva à perda progressiva da força muscular e, nos casos graves, à morte precoce. Aprovado para uso clínico em 2016 nos Estados Unidos e em 2017 no Brasil, o fármaco nusinersen – comercializado pelo laboratório norte-americano Biogen com o nome de Spinraza – melhorou a habilidade motora de 40% das crianças tratadas, segundo dados publicados em 2017 na revista científica New England Journal of Medicine. O medicamento modifica o funcionamento de um gene e aumenta a produção da proteína SMN, essencial à sobrevivência das células da medula espinhal que transmitem os comandos do cérebro para os músculos.
Injetado sob as membranas que protegem a medula espinhal, o nusinersen é um dos medicamentos mais caros do mundo. Ao ser lançado, as seis doses aplicadas no primeiro ano de tratamento custavam US$ 750 mil nos Estados Unidos. A partir do segundo ano, o número de aplicações e o custo da terapia, que dura a vida toda, caem pela metade. No Brasil, o medicamento é oferecido pelo Sistema Único de Saúde (SUS) desde abril para os casos que se manifestam nos primeiros 6 meses de vida e, a partir de agora, também para os que iniciam depois disso – aqui nascem por ano de 300 a 400 crianças com AME.
O nusinersen integra uma nova classe de compostos. Esses medicamentos surgem como desdobramento do sequenciamento do genoma humano, que transformou a biologia molecular e foi tema frequente nas páginas de Pesquisa FAPESP em seus 20 anos de existência. A revista publicou ao menos 10 capas sobre os vários projetos genoma e seus resultados, além de dezenas de reportagens menores. A definição da ordem dos 3,3 bilhões de bases nitrogenadas (adenina, A; timina, T; citosina, C; e guanina, G) do genoma humano abriu caminho para análises mais rápidas e precisas dos seus genes, o que, por sua vez, aprimoraram e baratearam o diagnóstico de doenças genéticas. Também levaram a tratamentos inovadores, alguns com o potencial de cura. Essas novas terapias, no entanto, ainda permanecem de acesso limitado pelo custo exorbitante.
Hoje se conhecem 4.147 genes associados a 6.499 doenças
“O sequenciamento do genoma humano permitiu um avanço importante no diagnóstico das doenças raras”, afirma a geneticista Lygia da Veiga Pereira, da Universidade de São Paulo (USP). São doenças causadas por alterações em um único gene (monogênicas) e, em geral, graves. Isoladamente, cada doença acomete uma proporção que varia de uma em cada mil a uma em cada 100 mil pessoas. Somadas, atingem quase 6% da população mundial, proporção semelhante à afetada pelo diabetes (8,5%). Por volta de 2000, quando um consórcio público internacional de sequenciamento competia com a empresa liderada pelo geneticista norte-americano John Craig Venter para concluir a tarefa de ler e ordenar as letras químicas do genoma humano, eram conhecidas 1.900 doenças monogênicas. Hoje estão mapeadas alterações em 4.147 genes associadas a 6.499 enfermidades, segundo a base Online Mendelian Inheritance in Man (Omim).
O avanço nas técnicas de sequenciamento e a evolução da bioinformática permitiram comparar o genoma de indivíduos saudáveis com o de pessoas com diferentes enfermidades e identificar a causa das doenças monogênicas – algo que ainda não se viu para as enfermidades que envolvem vários genes (poligênicas) e são mais complexas. “Esse conhecimento foi essencial para melhorar a identificação e o tratamento, além da prevenção, feita por meio de aconselhamento genético das famílias”, explica a geneticista Mayana Zatz, coordenadora do Centro de Pesquisa sobre o Genoma Humano e Células-Tronco (CEGH-CEL) da USP, um Centro de Pesquisa, Inovação e Difusão (Cepid) financiado pela FAPESP. No CEGH-CEL, um único teste detecta alterações em genes associados a quase 6,7 mil doenças (neuromusculares, cânceres hereditários, autismo e outras).
Identificar a causa das doenças genéticas melhora a qualidade de vida por permitir ao médico selecionar os remédios mais eficientes para atenuar os sintomas e evitar os medicamentos que os agravam. Também ajuda a preparar familiares e cuidadores para a evolução da enfermidade. Há ainda um benefício imponderável, lembra a médica geneticista Iscia Lopes Cendes, coordenadora do Laboratório de Genética Molecular da Universidade Estadual de Campinas (Unicamp) e pesquisadora do Brainn, outro Cepid financiado pela FAPESP. “Os testes genéticos muitas vezes dão um diagnóstico definitivo para essas doenças graves e reduzem a angústia dos pais”, explica.
Quando a primeira versão do genoma humano foi publicada, em 2001, houve otimismo exagerado de muitos pesquisadores, sentimento que repercutiu nos meios de comunicação e despertou na população anseios difíceis de serem atendidos. Na ocasião, o geneticista norte-americano Francis Collins, à época diretor do Instituto Nacional de Pesquisa do Genoma Humano (NHGRI) dos Estados Unidos, que coordenou o consórcio público de sequenciamento, comparou o genoma a um livro que narraria a jornada de nossa espécie no tempo. E acrescentou: “É um livro de medicina transformador, com ideias que darão aos prestadores de serviços de saúde poderes imensos para tratar, prevenir e curar doenças”.
O tom hiperbólico contrastou com o comedimento dos artigos científicos relatando o feito – um publicado em 15 de fevereiro de 2001 na revista Nature pelo consórcio integrado por Collins e outro no dia 16, na Science, pela equipe de Venter. Ao falar para os pares, o grupo de Collins foi cauteloso. Afirmou que haveria consequências para a medicina no longo prazo e encerrou o artigo dizendo: “Devemos estabelecer expectativas realistas de que os benefícios mais importantes não serão obtidos da noite para o dia”.
Na Science, Venter e seus colaboradores escreveram: “A sequência é apenas o primeiro nível de entendimento do genoma. Todos os genes e seus elementos de controle devem ser identificados; suas funções, em conjunto ou isoladamente, definidas; as variações na sequência deveriam ser descritas no mundo todo; e a relação entre as variações no genoma e as características fenotípicas [observáveis] específicas, determinadas”.
A ciência, como eles sabiam, não é rápida. “Nesses quase 20 anos, muita coisa progrediu, mas ainda não alcançamos as aplicações que muitos imaginavam”, afirma Cendes.
No consultório
Os avanços nas tecnologias de sequenciamento e nas estratégias de análise de dados pela bioinformática foram essenciais para que a medicina, quase duas décadas mais tarde, começasse a utilizar os conhecimentos da genômica na prática clínica. “Só recentemente algumas áreas médicas passaram de uma postura contemplativa para outra mais ativa”, conta o neurologista infantil Fernando Kok, pesquisador da Faculdade de Medicina da USP (FM-USP) e diretor médico da Mendelics, empresa de diagnósticos genéticos personalizados. Para ele, deve surgir em breve uma onda de terapias gênicas, que serão de acesso restrito pelo custo. “Ampliar o acesso será um problema para os gestores da área da saúde”, alerta.
Um motor do progresso na genômica foi o aprimoramento da tecnologia de sequenciamento. Em meados dos anos 1970, quando Allan Maxam e Walter Gilbert, nos Estados Unidos, e Frederick Sanger e Alan Coulson, na Inglaterra, desenvolveram as duas primeiras estratégias de sequenciar o DNA, o processo era lento e trabalhoso – Gilbert e Sanger dividiram o Nobel de Química de 1980 com o bioquímico Paul Berg. Gastava-se um dia para identificar a ordem de algumas centenas de bases de DNA. Só uma década depois surgiram os aparelhos automatizados, que empregavam o método de Sanger e foram usados no Projeto Genoma Humano.
Mais precisa, essa técnica sequencia, a cada vez, apenas um trecho curto de DNA, de até 900 bases. Nela, são produzidas cópias com um número crescente (1, 2, 3…) de bases. Apenas uma base (A, C, T ou G) é acrescentada a cada cópia – a última base é sempre marcada com um corante fluorescente (verde para A; azul para C; vermelho para T; e amarelo para G). Terminada a produção das cópias, elas são separadas por tamanho. Como se conhece a última base de cada cópia, é possível restabelecer a sequência original. O método de Sanger é usado ainda hoje para sequenciar moléculas isoladas de DNA, embora tenha sido substituído na maior parte das aplicações por uma técnica mais rápida e barata, o sequenciamento de nova geração (NGS), que identifica a ordem das bases de milhões de moléculas simultaneamente. Além das duas, adotadas em laboratórios clínicos, há uma terceira técnica, usada em pesquisa: o sequenciamento em tempo real de molécula única (SMRT), no qual uma fonte de laser ilumina cada base marcada com um corante fluorescente à medida que ela é adicionada à fita de DNA que está sendo copiada.
O custo da empreitada baixou de US$ 100 milhões em 2001 para cerca de US$ 1 mil em 2015, segundo cálculos do NHGRI. Esse valor permanece estável, embora empresas trabalhem para reduzir o preço do sequenciamento do genoma ou, ao menos, do exoma, a parte que contém os 24 mil genes que codificam proteínas, para centenas de dólares.
“Foi preciso chegar ao ponto de as técnicas baratearem muito e nos tornarmos bons o suficiente na interpretação dos dados para tornar essa tecnologia disponível na prática médica”, conta Cendes. Um trabalho orientado por ela e pela médica geneticista Antonia Marques de Faria, também da Unicamp, ajudou a embasar a aprovação de março deste ano de incorporar um novo teste genético no SUS para diganosticar deficiência intelectual: o sequenciamento do exoma.
Com diferentes manifestações clínicas, a deficiência intelectual é considerada um conjunto de doenças raras de diagnóstico clínico difícil. Suas diversas formas, somadas, atingem de 1% a 2% da população e prejudicam, em diferentes graus, o aprendizado, a habilidade de interação social e a capacidade de autocuidado. O diagnóstico atual no SUS é feito por teste de cariótipo (análise dos cromossomos, as estruturas em que os genes estão empacotados) e por microarray, técnica que analisa repetições no genoma e ainda é pouco disponível. A primeira identifica a causa em 3% dos casos e a segunda, em até 20%. Já a análise de exoma funciona em quase 40% das vezes. Na relação entre custo e benefício, a opção pelo exoma parece compensar, segundo estudo realizado por Joana Prota, aluna de doutorado orientada pelas pesquisadoras da Unicamp.
Doenças comuns
Se a genômica fez avançar a determinação das causas das doenças raras, ainda deixa a desejar no que diz respeito às enfermidades mais comuns, como diabetes, problemas cardiovasculares, doenças psiquiátricas e muitas formas de câncer, importantes do ponto de vista de saúde pública por atingirem um número elevado de pessoas. São doenças complexas e multifatoriais: resultam da ação de dezenas a centenas de genes, que interagem entre si e com o ambiente. Por essa razão, até hoje não se encontrou um gene que, sozinho, desempenhe papel importante no surgimento da hipertensão arterial, problema que atinge cerca de um terço da população adulta no mundo – as formas decorrentes de alteração em um único gene são raras. O mesmo ocorre com diabetes, transtornos psiquiátricos e vários tipos de câncer.
Nas doenças complexas, a contribuição de cada gene é pequena. Só é possível quantificar o efeito de cada um comparando um número grande de genomas, como começa a ser feito na Inglaterra, nos Estados Unidos e na China, onde há projetos para sequenciar o material genético de até 1 milhão de pessoas. Ainda assim, o que se encontrar por lá pode valer apenas para as populações europeias ou asiáticas. Em um artigo publicado em março deste ano na revista Cell, o geneticista Giorgio Sirugo, da Universidade da Pensilvânia, e dois colaboradores dos Estados Unidos afirmam que os estudos de ampla associação do genoma, destinados a identificar variantes associadas a traços complexos ou ao risco de desenvolver doenças, estão concentrados em poucas populações: 52% foram realizados com europeus e 21% com asiáticos. Segundo os pesquisadores, estudar grupos de outras origens é importante porque “os padrões de variação genética entre populações podem afetar o risco de desenvolver doenças e a eficácia e a segurança dos tratamentos”.
No Brasil, ainda são raros os estudos de avalição genômica da população. No CEGH-CEL, a equipe de Zatz realizou a análise do exoma de aproximadamente 1.500 paulistas com mais de 60 anos, em busca de variações gênicas protetoras, e a Brazilian Initiative on Precision Medicine (Bipmed), coordenada por Cendes, foi pioneira no compartilhamento público dos dados genômicos de quase 900 indivíduos (350 deles saudáveis, representantes da população geral). No A.C.Camargo Cancer Center, em São Paulo, os pesquisadores sequenciaram recentemente o genoma de 300 pessoas com câncer de estômago. Na USP, Lygia Pereira atualmente planeja obter dados de centenas de milhares de genomas de brasileiros para caracterizar as variações genéticas da população.
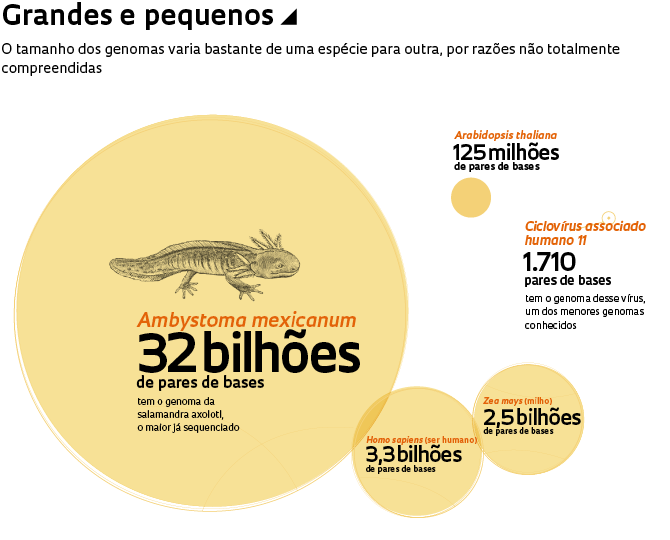
Até o momento, porém, as análises genômicas permitem, no máximo, associar a ocorrência de determinadas alterações genéticas ao risco (predisposição) de desenvolver um problema de saúde. “Para o diabetes e a obesidade, por exemplo, a contribuição desses estudos ainda é pequena, com potencial de, no médio prazo, permitir tratamentos mais efetivos”, comenta o endocrinologista especializado em doenças genéticas Alexander Jorge, da USP.
Apesar dessas limitações, as informações sobre alterações genéticas obtidas a partir do genoma e dos projetos que o seguiram têm auxiliado o diagnóstico e o tratamento de muitos dos quase 200 tipos conhecidos de câncer. “Na oncologia, as características genéticas dos tumores vêm sendo usadas para identificar o tipo de câncer e sua agressividade. Também permitem acompanhar a evolução da doença e a resposta ao tratamento”, relata a geneticista Anamaria Camargo, coordenadora do Centro de Oncologia Molecular do Instituto Sírio-Libanês de Ensino e Pesquisa (IEP), em São Paulo.
Assim como Camargo, muitos líderes dos principais centros de diagnóstico e tratamento oncológico do país acompanharam de perto o Projeto Genoma Humano e adquiriram conhecimentos de genômica ao participar dos primeiros projetos de sequenciamento do país, organizados e financiados pela FAPESP e por outras instituições. Em 1997, sob a coordenação dos bioquímicos Andrew Simpson e Fernando Reinach, à época, respectivamente, do Instituto Ludwig para Pesquisa sobre o Câncer (LICR) e da USP, e do geneticista Paulo Arruda e do bioinformata João Carlos Setubal, na Unicamp, equipes de 35 laboratórios paulistas iniciaram o sequenciamento do genoma da bactéria Xyllela fastidiosa, causadora da clorose variegada dos citros, ou amarelinho, doença que derrubava a produção dos laranjais paulistas.
“Foi um projeto concebido para capacitar os grupos para realizar sequenciamento de genomas, o que praticamente não existia no país”, afirma o físico José Fernando Perez, à época diretor científico da Fundação e atualmente diretor-presidente da Recepta Biopharma, empresa biotecnológica que desenvolve compostos para tratar câncer.
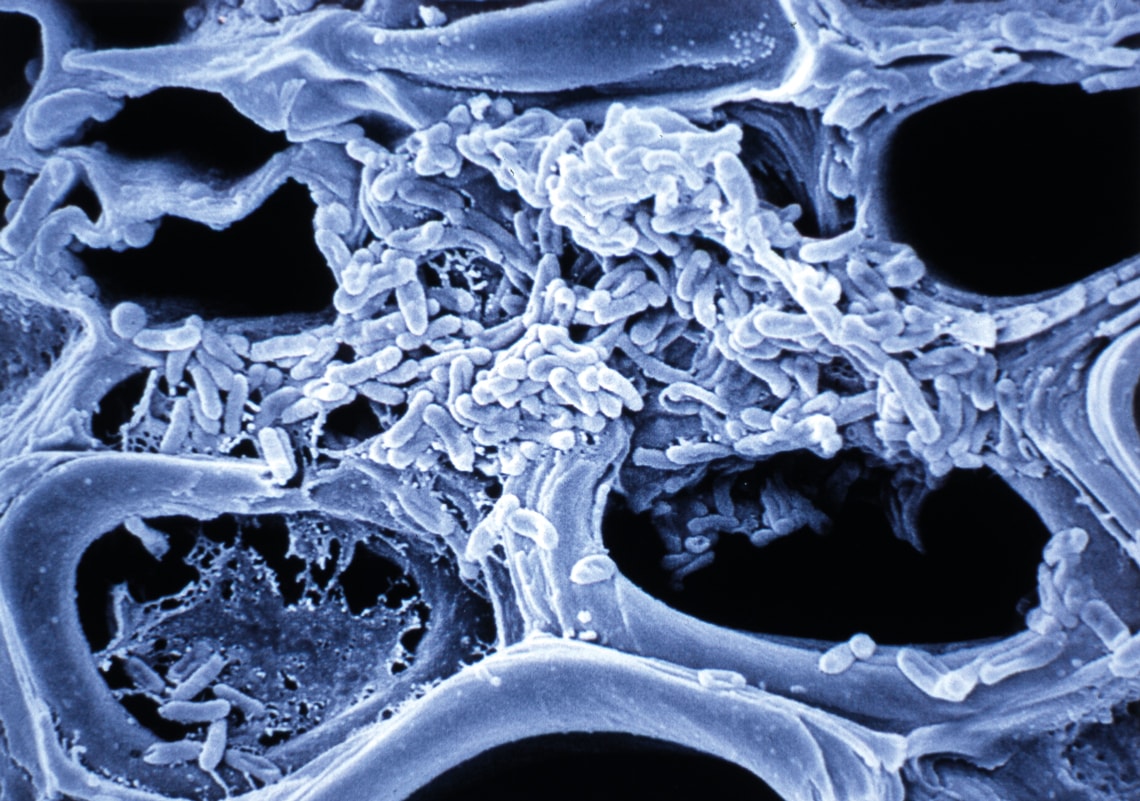
Vasos do caule de laranjeira bloqueados por colônia da bactéria Xyllela fastidiosa, o primeiro fitopatógeno a ter o genoma sequenciado
Cerca de três anos mais tarde, os 2,7 milhões de bases do genoma da bactéria haviam sido identificados e ordenados. O artigo mostrando o resultado foi capa da edição de 13 de julho de 2000 da revista Nature. Na época, o Projeto Genoma Humano ainda estava em curso, e o genoma de apenas oito organismos considerados modelos na biologia havia sido sequenciado: dois vírus, uma bactéria, uma levedura, um verme e uma planta. O genoma da Xyllela foi o primeiro de um organismo causador de doença em plantas, com relevância comercial. “Foi um momento em que o Brasil mostrou que, competindo em condições de igualdade, faz ciência de nível internacional”, afirma Simpson, atualmente diretor científico da Orygen Biotecnologia, empresa farmacêutica voltada para a produção de anticorpos, vacinas e outros medicamentos de origem biológica.
“Naquele período, o Brasil foi um dos raros países capazes de sequenciar o genoma completo de um organismo”, lembra Reinach, que há anos se desligou da universidade e hoje dirige um fundo de investimento em empresas inovadoras. De lá para cá, já se sequenciou o genoma de quase 19 mil organismos: 3,5 mil vírus; 14,7 mil bactérias; e 400 animais e plantas formados por uma ou mais células.
Durante a concepção do projeto da Xyllela, o oncologista Ricardo Brentani (1937-2011), então diretor da filial brasileira do LICR, decidiu organizar uma equipe e também participar do sequenciamento. “Brentani viu na Xyllela uma oportunidade de trazer a genômica para a oncologia”, conta Emmanuel Dias-Neto, coordenador do Laboratório de Genômica Médica do A.C.Camargo Cancer Center, do qual Brentani era também diretor. Ali, como no IEP, geneticistas e outros pesquisadores da área básica trabalham em colaboração com o corpo clínico do hospital usando informações genéticas dos tumores para orientar o tratamento e identificar o reaparecimento de tumores antes que se tornem detectáveis nos exames de imagem.
Em 1998, próximo à conclusão do genoma da Xyllela, alguns laboratórios que já participavam do projeto e outros que ainda não haviam entrado na onda genômica se organizaram para sequenciar, usando uma técnica desenvolvida por Dias-Neto e Simpson, trechos internos de genes que se encontram ativos nos tumores de mama, intestino, cabeça, pescoço, entre outros, com ênfase nos mais comuns na população brasileira. Os dados de 280 mil sequências foram depositados em um banco público de informações gênicas, o GenBank, e usados para auxiliar na identificação de genes nos cromossomos humanos sequenciados pelos grupos do Projeto Genoma Humano.

Reprodução
As reportagens de capa das edições nº 50, 51, 68 e 97 (
a partir da esq.) de Pesquisa FAPESP trataram de projetos ligados a sequenciamentos de genomas
ReproduçãoAo sequenciamento do genoma da Xyllela e do câncer, seguiu-se no Brasil o de outros patógenos de plantas (da bactéria Xanthomonas citri) e humanos (da bactéria Leptospira sp e do parasita Schistosoma mansoni), além do genoma do boi. Também se sequenciaram os genes expressos na cana-de-açúcar, o que possibilitou a produção de uma planta transgênica resistente a pragas e herbicidas, e os do eucalipto. Desse esforço, resultou ainda a criação de empresas de biotecnologia, como a Scylla, a Alellyx e a CanaVialis – as duas últimas foram compradas pela multinacional Monsanto e, depois, fechadas. Na visão de Perez, porém, “um dos legados mais importantes dos genomas coordenados pela Fundação foi o desenvolvimento da bioinformática no Brasil”.
Antes do início dos sequenciamentos em maior escala, o bioinformata tinha uma formação autodidata, conta João Meidanis, da Unicamp, que se graduou em matemática e optou pela bioinformática durante o doutorado nos Estados Unidos, quando se envolveu na análise do genoma da bactéria Escherichia coli. Desde então, surgiram cursos específicos para bioinformatas em algumas universidades brasileiras. “A comunidade cresceu, mas não no ritmo que se esperava e a bioinformática continua um gargalo para a análise das informações genômicas”, relata Meidanis, que também dirige a empresa Scylla Informática.
Arruda, da Unicamp, avalia a era dos sequenciamentos de genomas como um marco para a ciência brasileira. “Aprendemos a trabalhar em rede e a gerenciar grandes grupos de forma eficiente”, conta. “Também estabelecemos uma interação importante entre a universidade e empresas do setor privado.”
“Se não tivéssemos desenvolvido esses projetos naquele momento, hoje talvez não estivéssemos prontos para usar essa tecnologia que se tornou corriqueira”, conta a bióloga Marie-Anne van Sluys, da USP. Hoje ela coordena a participação brasileira em uma inciativa bem mais ambiciosa: o Earth Biogenome Project, que planeja sequenciar em 10 anos o genoma de todas as espécies de plantas e animais (uni ou pluricelulares) conhecidas. Será um trabalho hercúleo. São conhecidos cerca de 2,3 milhões de espécies, mas estima-se que, no total, sejam de 10 a 15 milhões.
Republicar