

GLAUCIA HAJJ/LICR
Príon celular em verde: abundante no neurônio…GLAUCIA HAJJ/LICREm maio de 1990 o ministro da Agricultura da Inglaterra, John Gummer, fez uma aparição pública desastrosa. Posou para fotógrafos e cinegrafistas saboreando um suculento hambúrguer ao lado de sua filha de 4 anos. Tinha a intenção de mostrar aos ingleses e ao resto do mundo que o consumo de carne bovina continuava seguro mesmo em meio à mais grave crise que a pecuária de seu país atravessava nos últimos tempos: a contaminação de parte do rebanho com a doença da vaca louca, a encefalopatia espongiforme bovina, que se espalhou pela Europa, pelos Estados Unidos e pelo Canadá e de 1987 até agora já obrigou a eliminação de 180 mil bois e vacas infectados.
Seis anos depois daquele hambúrguer os ingleses se lembrariam de Gummer e se sentiriam traídos quando começaram a surgir os primeiros casos da doença em seres humanos, provavelmente contraída pelo consumo de carne contaminada. A versão humana do mal da vaca louca era uma nova forma – a quarta conhecida – de uma enfermidade bastante rara e sem cura: a doença de Creutzfeldt-Jakob, que mata as células do sistema nervoso (neurônios) e deixa o cérebro cheio de buracos como uma esponja.
Descrita na Alemanha nos anos 1920 pelos neurologistas Hans Gerhard Creutzfeldt e Alfons Maria Jakob, essa enfermidade, que reduz o cérebro à metade de seu tamanho original, ganha nova explicação a partir de estudos recentes conduzidos no Brasil e no exterior. Em artigo publicado em abril na Physiological Reviews, o grupo de pesquisadores de São Paulo, Rio de Janeiro, Minas Gerais e Rio Grande do Sul coordenado pelo oncologista Ricardo Renzo Brentani, do Hospital A.C. Camargo, em São Paulo, apresenta a mais ampla revisão sobre os agentes infecciosos dessa doença, com informações que podem influenciar a terapia dessa enfermidade, que se instala sorrateiramente ao longo de 2 ou 3 décadas e evolui a uma velocidade assustadora, levando a uma morte trágica.
Os primeiros sinais surgem de forma sutil, como cansaço ou depressão. Em seguida, a falta de equilíbrio para caminhar ou manipular objetos aumenta progressivamente, os movimentos se tornam lentos e a visão embaralhada. Perde-se a fala, a memória para fatos recentes e fica cada vez mais difícil encontrar o caminho pelas ruas ou os objetos dentro de casa. “Em menos de 1 ano nove de cada dez pessoas infectadas se tornam debilitadas a ponto de não sair da cama e morrem”, afirma o neurologista Ricardo Nitrini, da Universidade de São Paulo (USP), que há 11 anos identificou o primeiro caso brasileiro de uma forma da doença causada por alteração genética.

GLAUCIA HAJJ/LICR
…e no astrócitoGLAUCIA HAJJ/LICRAlém da forma contraída pelo consumo de carne contaminada – a chamada nova variante de Creutzfeldt-Jakob – e da versão genética, passada de pais para filhos, há ainda outros dois tipos dessa doença que corrói o sistema nervoso central. O mais comum, dito espontâneo, surge ao acaso por razões desconhecidas e atinge uma pessoa em cada 1 milhão. O quarto tipo é transmitido pelo uso de equipamentos infectados em cirurgias, por transfusão sangüínea e até anos atrás também pela aplicação de hormônio do crescimento produzido a partir de cérebros de cadáver, hoje substituído pelo hormônio sintético para tratar distúrbios de crescimento.
O avanço da vaca louca nos pastos da Europa e da América do Norte e o surgimento da nova forma da doença em humanos – desde 1996 a nova variante de Creutzfeldt-Jakob matou na Inglaterra e nos países vizinhos 160 pessoas, entre elas a filha de um amigo do ex-ministro John Gummer – intensificaram a busca pela causa da enfermidade. O principal suspeito de provocar esse grupo de doenças desafiou por décadas os médicos e biólogos. Diferentemente do que acontece com outras doenças infecciosas, o causador da Creutzfeldt-Jakob não é, como se pensou por muito tempo, um vírus. Muito menos bactéria ou protozoário, microorganismos que se multiplicam por conta própria e são facilmente passados adiante. Hoje se acredita que uma proteína defeituosa conhecida como príon (sigla de partícula infecciosa proteinácea) provoque a doença. O simples contato do príon com uma proteína saudável encontrada em abundância na superfície dos neurônios a induziria a assumir a forma alterada, como uma pedra de dominó que tomba e derruba as demais da fileira sem que nada as possa deter. Mais estáveis que a proteína saudável, as moléculas deformadas aderem umas às outras, gerando longas fibras tóxicas para os neurônios.
A identificação do príon no cérebro de ovelhas com um tipo de encefalopatia espongiforme chamada scrapie e a explicação de como ele deformaria as proteínas normais renderam ao pesquisador norte-americano Stanley Prusiner o Nobel de Medicina de 1997 e levaram cientistas do mundo todo a investigar a proteína defeituosa e seus efeitos sobre o organismo. Enquanto só se tinham olhos para o príon, outra questão básica – e talvez mais importante – ecoava baixinho. O que fazia a proteína normal, a proteína príon celular, encontrada na superfície de todas as células do corpo e em maior quantidade no sistema nervoso central? Ninguém sabia, nem parecia se importar muito.
Até havia motivo para não se dar atenção ao príon celular. Por volta de 1990 o biólogo molecular Charles Weissmann criou uma linhagem de camundongos que não produziam essa proteína. Os animais não desenvolviam a doença espongiforme e aparentemente sobreviviam sem prejuízo à saúde. Por isso, acreditou-se que ela não desempenhasse papel importante no organismo. “Era uma visão limitada”, diz Brentani.

Reprodução The library of great painters/Bosh
A extração da pedra da loucura, de Hieronymus Bosch 1475-1480)Reprodução The library of great painters/BoshSuspeitando de que a natureza não desperdiçaria tempo nem energia para gerar uma proteína sem atividade biológica, Brentani apostou em sua intuição e seguiu contra a corrente. “Era a oportunidade de entrar em uma área de estudos quente pela qual ninguém havia se interessado”, conta. Uma carta publicada em 1991 na Nature estimulou-o a ir adiante. Três anos antes Brentani havia proposto uma teoria segundo a qual as duas fitas da molécula de ácido desoxirribonucléico (DNA) conteriam a receita para a produção de proteínas – e não apenas uma delas, como se imaginava. Também afirmava que as proteínas codificadas por trechos complementares das fitas de DNA teriam papéis complementares: seriam capazes de interagir quimicamente e se encaixar uma na outra como uma chave na fechadura. Do ponto de vista evolutivo, fazia sentido que os trechos de DNA que codificam uma proteína e a que se liga a ela estivessem próximos, já que é maior a probabilidade de migrarem juntos para outra região do material genético caso ocorra seu reposicionamento. Mas essa era uma hipótese em que, segundo Brentani, ninguém acreditava – exceto ele, claro.
Até que surgiu a carta da Nature. Nela o pesquisador Dmitry Goldgaber, da Universidade Estadual de Nova York, Estados Unidos, descrevia como o príon celular deveria interagir com a água – uma das características químicas das proteínas – e afirmava que, se Brentani estivesse certo, o trecho do DNA complementar ao do gene do príon celular conteria informação sobre a proteína que possivelmente o acionaria. Era uma pista a não se desperdiçar.
Então estudioso de proteínas associadas ao câncer, Brentani resolveu analisar o príon e a molécula que funcionava como seu interruptor. Ele, a bioquímica Vilma Martins, do Instituto Ludwig de Pesquisa sobre o Câncer (LICR), e o bioquímico Vivaldo Moura Neto, da Universidade Federal do Rio de Janeiro (UFRJ), deduziram a estrutura dessa outra proteína e a descreveram em 1997 na Nature Medicine.
A proteína por eles apresentada – mais tarde identificada como STI-1, sigla de stress inducible protein 1 – era composta por 543 aminoácidos (os blocos formadores das proteínas) e quase duas vezes maior do que o príon celular. Faltava descobrir o que ambas faziam. “Tínhamos duas hipóteses: ou não serviam para nada ou eram fundamentais para fenômenos importantes para os neurônios, como o processo de neuritogênese [formação das ramificações que conectam os neurônios entre si]”, comenta Brentani.
UCSF
Uma proteína, duas formas: o príon celular…UCSFComo neurônios não era a especialidade do grupo, ele e Vilma convidaram o neurocientista Rafael Linden, do Instituto de Biofísica da UFRJ, para colaborar nos testes seguintes. O complexo formado pelo príon celular e a STI-1 se mostrou essencial tanto para o amadurecimento e a formação dos prolongamentos dos neurônios como para protegê-los da morte celular programada, a apoptose (ver Pesquisa FAPESP nº 94).
Mas essas não eram as únicas funções da dupla. Experimentos com camundongos feitos em parceria com Iván Izquierdo, um dos mais respeitados estudiosos de memória no mundo e atualmente pesquisador da Pontifícia Universidade Católica do Rio Grande do Sul (PUC-RS), revelaram que o príon celular e a STI-1 são fundamentais para a formação da memória. Sem eles, os animais têm dificuldade de lembrar algo que aprenderam horas antes (memória de curto prazo) e também dias atrás (memória de longo prazo). Testes com camundongos geneticamente alterados para não produzir o príon celular, como os criados por Charles Weissmann, comprovaram que esses animais só eram aparentemente normais. Quando envelheciam, apresentavam mais dificuldades de memória do que os camundongos que fabricavam o príon celular.
O grupo brasileiro também viu que a forma saudável do príon gera efeitos distintos em tecidos diferentes. Na UFRJ a equipe de Linden constatou que essa proteína modula a resposta do sistema imunológico às inflamações, ora aumentando, ora reduzindo a atividade das células de defesa. O príon celular estimula a ação dos neutrófilos, as células de defesa mais abundantes no organismo. Produzidas a uma quantidade de 100 bilhões por dia no interior dos ossos longos, são as primeiras a chegar ao local da inflamação, onde rapidamente englobam e destroem microorganismos invasores como bactérias. Quando Linden provocava uma inflamação em camundongos, observou que os animais geneticamente alterados para não produzir o príon celular apresentavam um número menor de neutrófilos, que também eram mais lentos do que os roedores normais. Um efeito nada desejável no caso de uma infecção.
Verificou-se o efeito oposto com os macrófagos, células do sistema de defesa que atuam como uma espécie de lixeiro, eliminando células mortas. Camundongos sem o príon celular tinham macrófagos mais ativos do que os animais que fabricavam a proteína, um resultado que nem sempre favorece os animais geneticamente alterados, pois a ação exagerada dos macrófagos pode causar lesões nos tecidos saudáveis. “A resposta à inflamação e à presença de células mortas depende de um ajuste fino”, explica Linden. “Não é desejável que sejam ausentes nem exacerbadas. Sem resposta inflamatória o corpo não resiste a infecções, mas inflamação em excesso também pode matar”.

UCSF
… e sua forma infecciosaUCSFTambém há evidências de que o príon celular protege as células do coração contra a agressão química. No Hospital A.C. Camargo, Vilma e a médica Beatriz de Camargo analisaram a presença de uma forma ligeiramente alterada (variante) das proteínas príon celular em 160 pacientes tratados na infância com adriamicina, medicamento que pode causar lesões cardíacas. Dados preliminares sugerem que os portadores da forma variante do príon celular eram mais suscetíveis aos efeitos tóxicos do composto do que aqueles com a versão normal da proteína.
À medida que os resultados brotavam no laboratório, tornava-se evidente que o príon celular era fundamental para manter o organismo saudável, nada mau para uma molécula que até poucos anos atrás era considerada sem importância biológica. Mas ainda não se compreendia por que, em determinadas situações, ela protegia e em outras danificava os tecidos. Um passo importante era saber como essa proteína em forma de balão de festa com um barbante pendurado, que fica na superfície externa das células, se comunicava com o interior.
Vilma, Brentani e Linden recorreram então à ajuda do biólogo celular Marco Antonio Prado, da Universidade Federal de Minas Gerais (UFMG), que investiga o transporte de moléculas no interior das células. Em parceria com Vilma e Kil Sun Lee, do Ludwig, Prado e Ana Maria Magalhães marcaram o príon celular de neurônios com um corante verde fluorescente para acompanhar o caminho que percorria e levaram as células ao microscópio confocal, que permite observá-las vivas. Em seguida, com o auxílio de Byron Caughey, dos Institutos Nacionais de Saúde dos Estados Unidos, marcaram o príon infeccioso e viram sua entrada nos neurônios (ver Pesquisa FAPESP nº 115).
Ancorado em regiões mais espessas da superfície celular por uma longa molécula de açúcar e lipídios em forma de barbante, o príon celular desliza para áreas mais delgadas da membrana dos neurônios. Ali é tragado para o interior de vesículas contendo ácidos, onde se conecta a outras proteínas e envia comandos para o núcleo ou outras regiões. Do início do mergulho até a emersão na superfície, o príon celular não gasta mais do que 1 hora e meia.
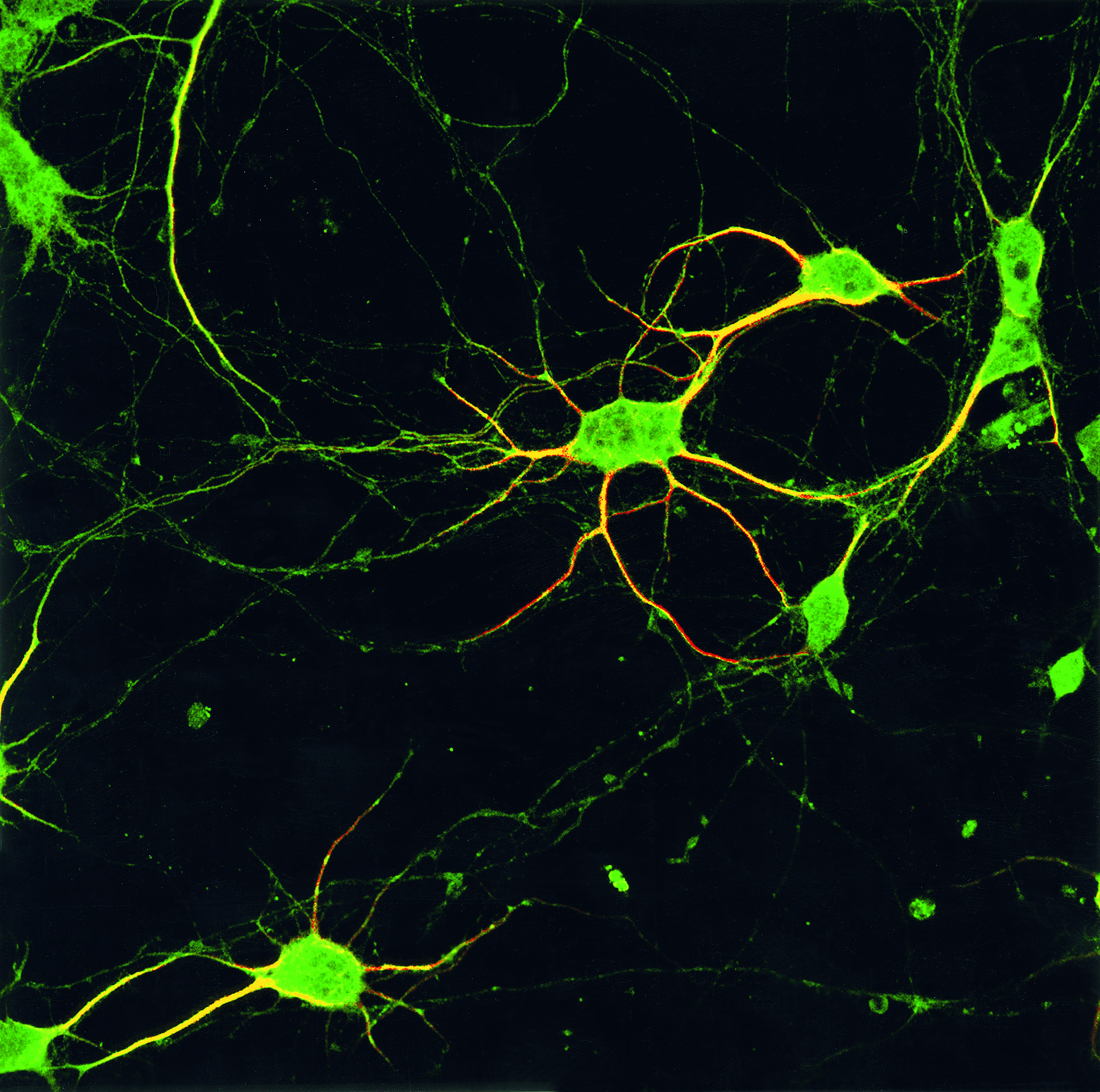
GLAUCIA HAJJ/LICR
Rede de neurônios: príon celular (verde) favorece a conexão entre as célulasGLAUCIA HAJJ/LICRNão é um deslocamento ao acaso, como constatou o grupo brasileiro. O príon celular só se move na superfície dos neurônios depois que proteínas específicas se acoplam a ele, ativando-o. Como um anfitrião que recebe os convidados em uma festa, o príon saudável conduz outras proteínas para o interior dos neurônios. Uma vez no interior da célula, o complexo formado pelo príon e sua proteína ativadora envia sinais químicos que ordenam a emissão de prolongamentos ou a produção de compostos que protegem o neurônio da morte, detalham os pesquisadores em um artigo a ser publicado nos próximos meses no Journal of Neuroscience. “Sem esse mergulho no interior da célula a comunicação mediada pelo príon celular fica truncada”, diz Linden.
Quanto mais se descobria sobre o príon celular, mais dúvidas surgiam. No final de 2006 Linden, Vilma, Prado, Izquierdo e Brentani começaram a rever tudo o que havia sido publicado sobre o príon saudável e o defeituoso com o objetivo de chegar a um quadro geral mais claro. Da análise de 597 artigos, emergiu a mais ampla revisão sobre o tema, publicada em abril na revista Physiological Reviews, com uma visão unificada sobre o funcionamento do príon celular e uma nova interpretação de como surgem enfermidades como a doença de Creutzfeldt-Jakob e o mal da vaca louca.
No trabalho intitulado “Physiology of the prion protein”, as equipes de São Paulo, Rio, Minas e Rio Grande do Sul propõem que o príon celular funcione como um ímã seletivo ao qual só aderem certas moléculas encontradas no organismo. A STI-1, claro, não é a única. Estudos feitos no Brasil e no exterior identificaram outras 30 proteínas que se ligam ao príon celular, acionando diferentes cascatas de reações químicas que representam comandos celulares distintos. “Acreditamos que o príon celular ajude a organizar os sinais do exterior antes de serem enviados para o interior das células”, diz Prado.
Segundo Linden, esse papel de ímã seletivo ou plataforma de montagem de complexos de sinalização permite explicar resultados experimentais até então contraditórios, como a proteção contra a morte celular em determinadas situações e ou a ação tóxica em outras. “Essa atividade de plataforma de montagem de sinais químicos é tão essencial para a vida que possivelmente outras proteínas desempenhem o mesmo papel no organismo”, diz Brentani. “Por essa razão os camundongos geneticamente alterados para não produzir o príon celular sobrevivem aparentemente sem prejuízo”, explica.
Esse novo papel altera a compreensão de como se instalam as doenças causadas por príons. De acordo com a nova interpretação, na doença de Creutzfeldt-Jakob os neurônios não morreriam apenas porque a adesão dos príons infecciosos gera aglomerados tóxicos. O grupo brasileiro aposta que a morte celular ocorra também pela perda de moléculas saudáveis de príon, que deixaria os neurônios desprotegidos contra agressões químicas. Segundo Prado, é possível que o efeito tóxico do príon infeccioso se intensifique com a perda do príon celular. “Só saberemos se estamos certos à medida que as idéias apresentadas nesse trabalho começarem a ser testadas”, diz Linden.
A expectativa é que a compreensão de como funciona o príon celular leve a alternativas de tratamento para doenças causadas por príons e enfermidades neurodegenerativas como o mal de Alzheimer, associado à aglomeração de uma proteína cuja produção é controlada pelo príon saudável. “As abordagens terapêuticas que se basearam exclusivamente no que se conhece sobre a forma defeituosa do príon não produziram bons resultados”, conta Linden. Um medicamento usado na década de 1930 contra a malária, a quinacrina, havia se mostrado capaz de impedir a agregação do príon infeccioso nos experimentos com neurônios in vitro. Mas não impediu o avanço da enfermidade quando testado em seres humanos. “Até o momento, não há tratamento eficaz”, afirma Ricardo Nitrini, da USP.
Com Hélio Gomes e Sérgio Rosemberg, da USP, e Leila Chimelli, da UFRJ, Nitrini e Vilma integram a equipe responsável no país pelo diagnóstico de doenças causadas por príon, cuja notificação é obrigatória desde 2005. É uma medida fundamental para conhecer as regiões mais afetadas e as populações mais suscetíveis às quatro formas da doença de Creutzfeldt-Jakob. De 2005 a 2007, o grupo analisou 35 casos suspeitos, dos quais 26 foram considerados prováveis – a confirmação é feita pela análise do tecido cerebral após a morte. Eram pessoas que haviam desenvolvido a doença espontaneamente. Nenhum caso surgiu pelo consumo de carne infectada. “Certamente há subnotificação da doença no país, onde se espera que surjam até 200 casos por ano”, diz Vilma.
Em paralelo, a equipe de Vilma no Ludwig segue com os estudos sobre a ação da STI-1. Nos últimos anos, o grupo constatou que um fragmento dessa molécula, um peptídeo de 16 aminoácidos, desempenha a mesma função que a proteína inteira e favorece a formação da memória em camundongos. Testes iniciais com células em uma placa de vidro também sugerem que o peptídeo impeça o desenvolvimento de um tumor cerebral agressivo, o glioblastoma, que mata em 6 meses, razão por que esse trecho da molécula foi patenteado pelo Ludwig em 2007 nos Estados Unidos. “São dados promissores”, afirma Vilma. Por enquanto, não se pode dizer mais do que isso até que sejam feitos testes com animais de laboratório e, se tudo der certo, em seres humanos.
O Projeto
Papel da proteína príon celular em processos fisiológicos e patológicos II (03/13189-2); Modalidade Projeto Temático; Coordenadora Vilma Regina Martins – Instituto Ludwig; Investimento R$ 2.053.618,66