 O projeto Genoma Humano do Câncer – iniciado em março do ano passado com financiamento da FAPESP em parceria com o Instituto Ludwig de Pesquisas sobre o Câncer – vai dobrar o número de seqüências de regiões importantes do genoma humano que pretende identificar. Estipulada inicialmente em 500 mil até abril de 2001, a meta agora é atingir 1 milhão, em um prazo menor – até o final deste ano. Para chegar lá, os 30 laboratórios em operação devem ganhar reforços, tanto na equipe, hoje de 150 pesquisadores, quanto nos equipamentos, e outros centros de seqüenciamento podem ser incorporados ao projeto. Pretende-se também tornar ainda mais eficiente a identificação de genes, uma etapa crucial do trabalho, realizada nos centros de bioinformática.
O projeto Genoma Humano do Câncer – iniciado em março do ano passado com financiamento da FAPESP em parceria com o Instituto Ludwig de Pesquisas sobre o Câncer – vai dobrar o número de seqüências de regiões importantes do genoma humano que pretende identificar. Estipulada inicialmente em 500 mil até abril de 2001, a meta agora é atingir 1 milhão, em um prazo menor – até o final deste ano. Para chegar lá, os 30 laboratórios em operação devem ganhar reforços, tanto na equipe, hoje de 150 pesquisadores, quanto nos equipamentos, e outros centros de seqüenciamento podem ser incorporados ao projeto. Pretende-se também tornar ainda mais eficiente a identificação de genes, uma etapa crucial do trabalho, realizada nos centros de bioinformática.
O Genoma Humano do Câncer deve ganhar também as primeiras aplicações práticas, por meio do subprojeto Genoma Clínico do Câncer, sujeito aos ajustes finais nas próximas semanas. Essa nova vertente do trabalho vai se abrir à participação de médicos cujo conhecimento possa contribuir com o desenvolvimento de diagnósticos precoces e novos procedimentos terapêuticos, a partir das informações já disponíveis. De todo modo, os planos devem dobrar o orçamento do projeto, para o qual a FAPESP e o Instituto Ludwig destinaram cerca de US$ 10 milhões. “É preciso responder à percepção de que há uma nítida oportunidade para dar uma contribuição relevante para o Genoma Humano e para o estudo do câncer”, comenta José Fernando Perez, diretor científico da FAPESP.
A expansão do projeto apóia-se em resultados. Sob a coordenação do Instituto Ludwig, os pesquisadores atrelaram-se a um ritmo de trabalho altamente produtivo, que fez com que as metas fossem ultrapassadas. Até o início de maio, o grupo brasileiro deverá ter acumulado informações a respeito de cerca de 300 mil seqüências do genoma humano – os laboratórios produzem cerca de 2.500 novas seqüências por dia. Se, por uma fatalidade, o trabalho tivesse de parar no estágio em que se encontra, o Brasil já desfrutaria uma posição de destaque entre os primeiros grupos de estudo do genoma humano. Ainda não há um método consensual que peneire os genes dessa massa de informações, mas calcula-se que possa haver cerca de 45 mil genes no material já analisado, algo próximo da metade dos estimados 100 mil genes do genoma humano.
Câncer de mama
Estima-se que um terço das informações processadas sejam mundialmente inéditas. Portanto, há um material passível de patenteamento, que poderia assegurar a autonomia no desenvolvimento de novos diagnósticos ou medicamentos. No dia 6 de abril, seguiu para a filial do Ludwig em Londres a documentação necessária para a solicitação de patenteamento internacional do primeiro gene resultante desse projeto, a RNA helicase, descoberto no início de 1999, pouco antes da constituição do projeto conjunto coma FAPESP. Os autores do pedido de patente – o bioquímico Andrew Simpson, coordenador do projeto, e os biólogos Emmanuel Dias Neto e Anamaria Camargo – já haviam verificado que a RNA helicase tem uma expressão intensa em câncer de mama.
Este ano, comprovaram que ele é único, sem similaridade com qualquer outro gene humano. É, sim, muito semelhante a um da mosca-de-fruta, a Drosophila melanogaster, cujo genoma completo – com 160 milhões de pares de bases e estimados 13.600 genes – foi apresentado no final de março por um grupo de pesquisadores do Instituto Médico de Howard Hughes e da empresa Celera Genomics, dos Estados Unidos. O acúmulo de evidências motivou os pesquisadores do Ludwig em São Paulo a trabalhar até chegar à seqüência completa do gene, com 1.889 nucleotídeos.
“Estou realmente entusiasmado com os resultados”, observa Marcelo Bento Soares, pesquisador da Universidade de Iowa, dos Estados Unidos, e um dos integrantes do Steering Commitee, o comitê externo de avaliação. Soares, que esteve em fevereiro no Brasil, aponta cinco benefícios estratégicos propiciados pelo projeto brasileiro. Primeiro, a quantidade de dados relevantes para a comunidade científica mundial, “de modo como só os grandes projetos internacionais proporcionam”. Segundo, o fato de o grupo “ter se proposto metas ambiciosas, que estão excedendo”. O terceiro ponto que ele destaca é a aproximação entre o Ludwig Brasil e internacional e a comunidade científica nacional, por meio da FAPESP. O quarto: a demonstração de que é possível coordenar grupos de instituições distintas, de modo bastante produtivo.
E, por fim, o fato de a FAPESP ter tomado a liderança e proposto o desenvolvimento do programa, como já faz o National Institute of Health dos Estados Unidos, em vez de esperar a demanda dos cientistas. “Em breve, quando as seqüências do projeto brasileiro forem colocadas inteiramente à disposição da comunidade científica internacional, o impacto será grande”, antecipa Soares. Não deve tardar. Já estão em domínio público cerca de 65 mil seqüências e até junho o Ludwig deve completar 250 mil seqüências depositadas nos bancos internacionais de DNA, o que tornará a informação obtida no Brasil disponível para os centros de pesquisa do mundo todo.
Essa providência não foi tomada antes – a despeito do compromisso inicial entre as instituições de pôr tudo no domínio público (uma posição reiterada em março pelo presidente dos Estados Unidos, Bill Clinton, e semanas depois pelos cientistas da Academia Nacional de Ciências do Estados Unidos e da Real Sociedade de Londres) – porque se pretendia analisar um pouco melhor as descobertas.
 Achados no cromossomo 22
Achados no cromossomo 22
A equipe que trabalha em São Paulo não apenas acompanha a corrida mundial do genoma humano, mas também tem encontrado novidades até mesmo em espaços percorridos por grupos de pesquisas internacionais. É o caso do cromossomo 22, o segundo menor entre os 23 do genoma humano (o menor de todos é o 21). O mapeamento do cromossomo 22 – apresentado com alarde por representar o resultado inaugural do Projeto Genoma Humano, um consórcio internacional de centros de pesquisa que pretende identificar os 23 cromossomos do genoma humano até 2002 – ganhou as páginas da revista Nature do dia 2 de dezembro de 1999. Assinado por 217 pesquisadores dos Estados Unidos, Canadá, Reino Unido e Japão, o artigo descreve 545 genes, quase a metade dos estimados 1.000 desse cromossomo.
O biólogo Sandro José de Souza, coordenador de bioinformática do Genoma Humano do Câncer, soube da preparação do artigo algumas semanas antes de sua publicação. Ao perceber que o cromossomo 22 era uma oportunidade para pôr em prática a metodologia empregada no projeto paulista, foi à luta. Chamou sua equipe do Ludwig e planejou o trabalho, que terminou ainda em dezembro, de modo gratificante. “Conseguimos identificar cerca de 100 genes que o resto do mundo não descobriu”, festeja o pesquisador. Louve-se também o aproveitamento do material disponível. Segundo Souza, o reconhecimento de genes do cromossomo 22 baseou-se em apenas 100 mil seqüências, 15 vezes menor do que a base de dados sobre a qual trabalhou a equipe do artigo da Nature.
Não se trata de seguir à sombra ou de ir contra o mundo – tanto que já se tem como certo que o projeto paulista, daqui para a frente, seguirá uma trilha internacional, a da busca e interpretação apenas dos genes transcritos ativos e de suas variantes, os chamados transcriptons. É uma forma de concentrar o trabalho nos trechos relevantes dos genes, os chamados exons, sem incluir as partes apenas estruturais, os íntrons, que não levam à formação de proteínas. Devem, portanto, residir nos transcriptons as informações mais importantes para entender a genética do câncer. “Interpretar os transcriptons é o atual desafio do mundo inteiro”, diz Simpson.
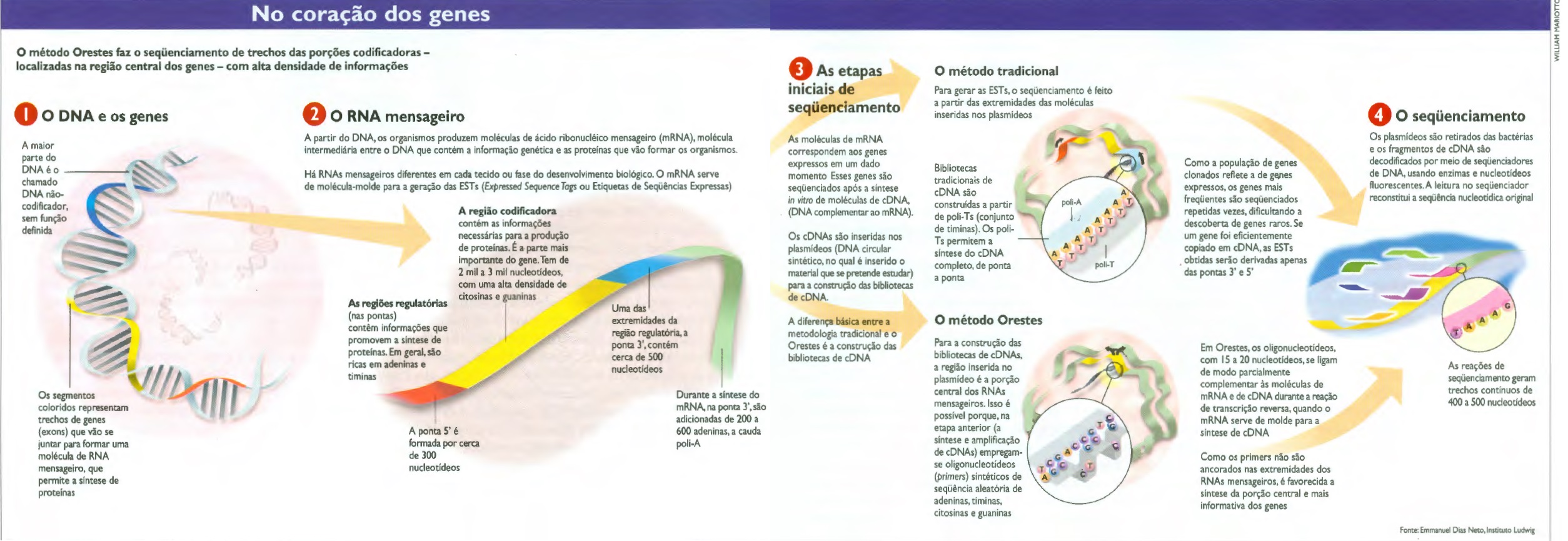
No início de abril, ao encontrar-se com os coordenadores dos laboratórios para tratar das possibilidades de divulgação dos resultados obtidos em publicações científicas, Simpson comentou que estava orgulhoso não apenas pelas descobertas, mas também pela maneira como estão trabalhando. A satisfação e os resultados obtidos até o momento devem-se ao emprego de uma técnica inédita de seqüenciamento, o Orestes, sigla de Open Reading Frames EST Sequences. Criado por Simpson e pelo biólogo Emmanuel Dias Neto, aperfeiçoado nos últimos oito anos e amadurecido ao longo do Genoma Xylella, o Orestes busca genes expressos (ativos), os transcriptons – uma abordagem mais do que oportuna quando não se pensa mais em investigar o genoma inteiro, mas apenas esses trechos ativos dos genes.
Abordagem incomum
Em vez de ler o gene a partir das pontas, como os métodos em uso em outros países, o método Orestes concentra-se nas informações que se encontram na região central da molécula de RNA mensageiro e, desse modo, tem mais condições de encontrar genes raros. É uma busca insana. Em cada célula, conta Dias Neto, há em média 11.000 genes raros, cada um copiado apenas dez vezes. No extremo oposto, há menos de dez genes abundantes, ainda que possa haver cerca de 12.000 duplicatas de cada um deles. Em mil genes, há 99,9% de chance de encontrar um gene abundante e 0,00001% de achar um raro. E há casos em que as funções mais importantes para o organismo são cumpridas pelos raros, que às vezes só agem em combinação com outro, também raro.
Para complicar um pouco, a parte codificadora de um gene qualquer geralmente está dividida em diversos pedaços ao longo da molécula de DNA – os exons. O mecanismo de splicing, descoberto em 1977 pelo químico norte-americano Phillip Sharp, prêmio Nobel de 1993, que em setembro esteve no Brasil (ver Notícias Fapesp nº 46), junta os exons. Souza conta que, por um processo de splicing alternativo, é possível que mais de uma proteína se forme a partir do mesmo gene com a inclusão ou exclusão de um ou mais exons. “O método Orestes contribui para a identificação dessas variantes de splicing “, conta Souza. Uma dessas variações foi encontrada no cromossomo 22: um transcrito do gene fosfatidil-4-kinase, responsável pela formação de uma enzima que adiciona um átomo de fósforo a outras proteínas, foi encontrado com um exon a menos do que a forma conhecida.
Ao contribuir no desenho do genoma humano e acenar com uma melhor compreensão dos mecanismos genéticos do câncer, o Orestes tem conquistado o respeito internacional e feito Simpson viver dias de emoção intensa. No início de fevereiro, ele conta, havia sido muito bem recebida sua apresentação sobre as descobertas brasileiras do cromossomo 22 no Genomics Two Thousands, um encontro realizado na Flórida, Estados Unidos, que tratou dos avanços na pesquisa de genomas humanas e de bactérias.
No final de abril, outra dose de contentamento. No dia 28, uma sexta-feira, saiu o artigo publicado na revista Proceedings of The National Academy of Sciences, a PNAS, dos Estados Unidos, por indicação do próprio Phillip Sharp. Para completar, no dia 29 a equipe de Simpson chegaria à marca de 250 mil seqüências identificadas. “Sinto que o cenário internacional do genoma está mudando, a nosso favor”, comenta Simpson.
A corrida pelo mapeamento
Ainda não se pode dizer se o geneticista Craig Venter, presidente da empresa norte-americana Celera Genomics, entrou efetivamente para a história ao anunciar, no dia 6 de abril, a conclusão da primeira etapa do seqüenciamento do genoma humano: a leitura dos 3,5 bilhões de pares de nucleotídeos ou bases (adenina, timina, citosina e guanina) do genoma humano.
A notícia causou impacto no mundo todo, por indicar que a iniciativa privada poderia vencer a corrida para o seqüenciamento do genoma humano, da qual participam, principalmente, instituições públicas de pesquisa. Mas ainda falta muito trabalho para identificar, nas bases já listadas, os cerca de 100 mil genes humanos, cada um deles com um arranjo único de cerca de 3 mil nucleotídeos. Os genes ocupam apenas 3% da molécula de DNA, constituído quase integralmente por regiões sem funções definidas, prováveis resquícios evolutivos do genoma humano.
Segundo os especialistas, o que Venter fez até agora pode ser comparado a uma fotografia na qual não se distinguem pessoas, bonecos, rochas ou árvores. Ele teria agora de distinguir cada elemento da foto e, mais ainda, descobrir a profissão de cada pessoa. Mesmo assim, Craig, que trabalhou com base no genoma de uma pessoa, diz que tem condições de concluir a segunda parte do trabalho dentro de dois meses, por meio de análises feitas por computadores de alto desempenho. Há alguns anos, ele compete no seqüenciamento genético humano com o Projeto Genoma Humano, um consórcio público que conta com financiamento público, já seqüenciou cerca de 2 bilhões de pares de bases e pretende anunciar em maio um rascunho completo do genoma humano.
Num encontro de cientistas no Canadá, na semana seguinte, Francis Collins, do Instituto Nacional de Pesquisa sobre o Genoma Humano, dos Estados Unidos, reconheceu o esforço da Celera, mas lembrou que o trabalho foi feito com apenas três – e não dez, conforme os procedimentos científicos internacionalmente reconhecidos – releituras da seqüência de bases. Segundo ele, “ninguém vai conseguir completar o seqüenciamento nos próximos dois anos”.
Republicar