Em um ano de pandemia, oficialmente decretada em 11 de março de 2020 pela Organização Mundial da Saúde (OMS), o mundo assistiu, atônito, ao adoecimento de 122 milhões de pessoas e à morte de ao menos 2,7 milhões – um terço delas ocorridas em apenas três países: Estados Unidos, Brasil e México. Também pôde acompanhar, em tempo quase real e com um nível de detalhe talvez nunca visto antes, a evolução do patógeno que pôs de joelhos o sistema de saúde e afetou profundamente a economia de muitas nações. Desde que foi identificado no final de 2019 na cidade de Wuhan, na região central da China, o vírus Sars-CoV-2 sofreu uma série de transformações enquanto se espalhava pelo planeta. À medida que infectava mais e mais pessoas e se replicava, acumulou pequenas modificações em seu material genético até que, em diferentes locais e momentos, já estava tão diferente do original que passou a ser considerado uma nova variante, que, ao prosperar e se disseminar, começa a ser chamada de linhagem – a linhagem agrupa exemplares com a mesma origem e muito semelhantes entre si, mas que podem possuir pequenas diferenças.
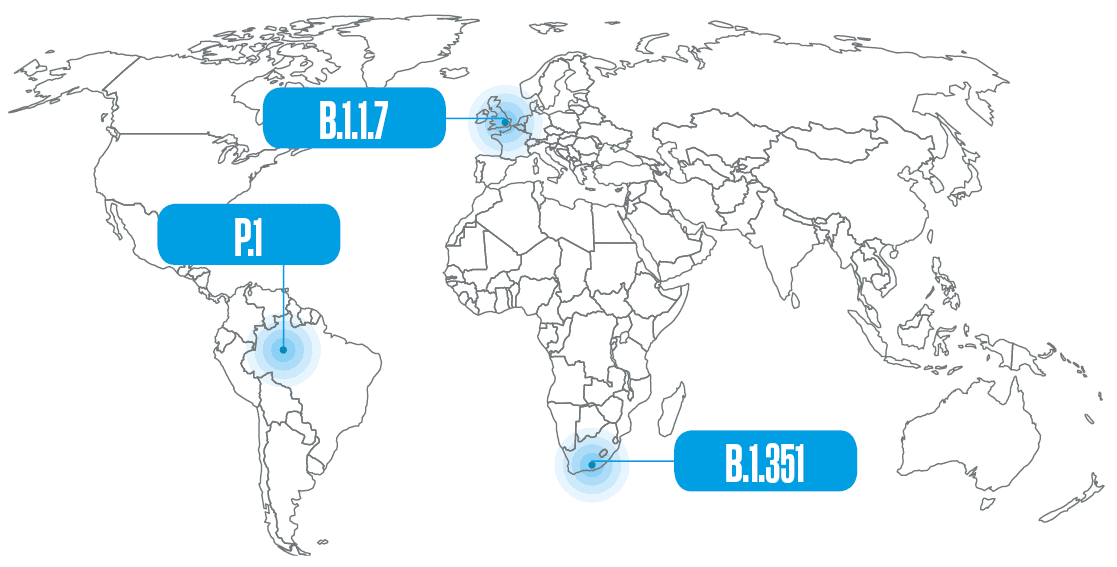
Em 15 de março deste ano, a Nextstrain, uma ferramenta de visualização de genomas virais, listava nada menos do que 359 linhagens do novo coronavírus catalogadas desde dezembro de 2019 pelo sistema de classificação Pangolin. Entre tantas, três delas – uma surgida no Reino Unido, outra na África do Sul e uma terceira no Brasil – vêm causando especial preocupação por serem mais transmissíveis, poderem escapar à ação de anticorpos e, em alguns casos, provocarem doença mais grave do que as que circulavam anteriormente. “Com as ferramentas genéticas de que dispomos atualmente, estamos assistindo à evolução desse patógeno ao mesmo tempo que ela ocorre”, afirma o virologista José Luiz Proença Módena, coordenador do Laboratório de Estudos de Vírus Emergentes da Universidade Estadual de Campinas (Unicamp).
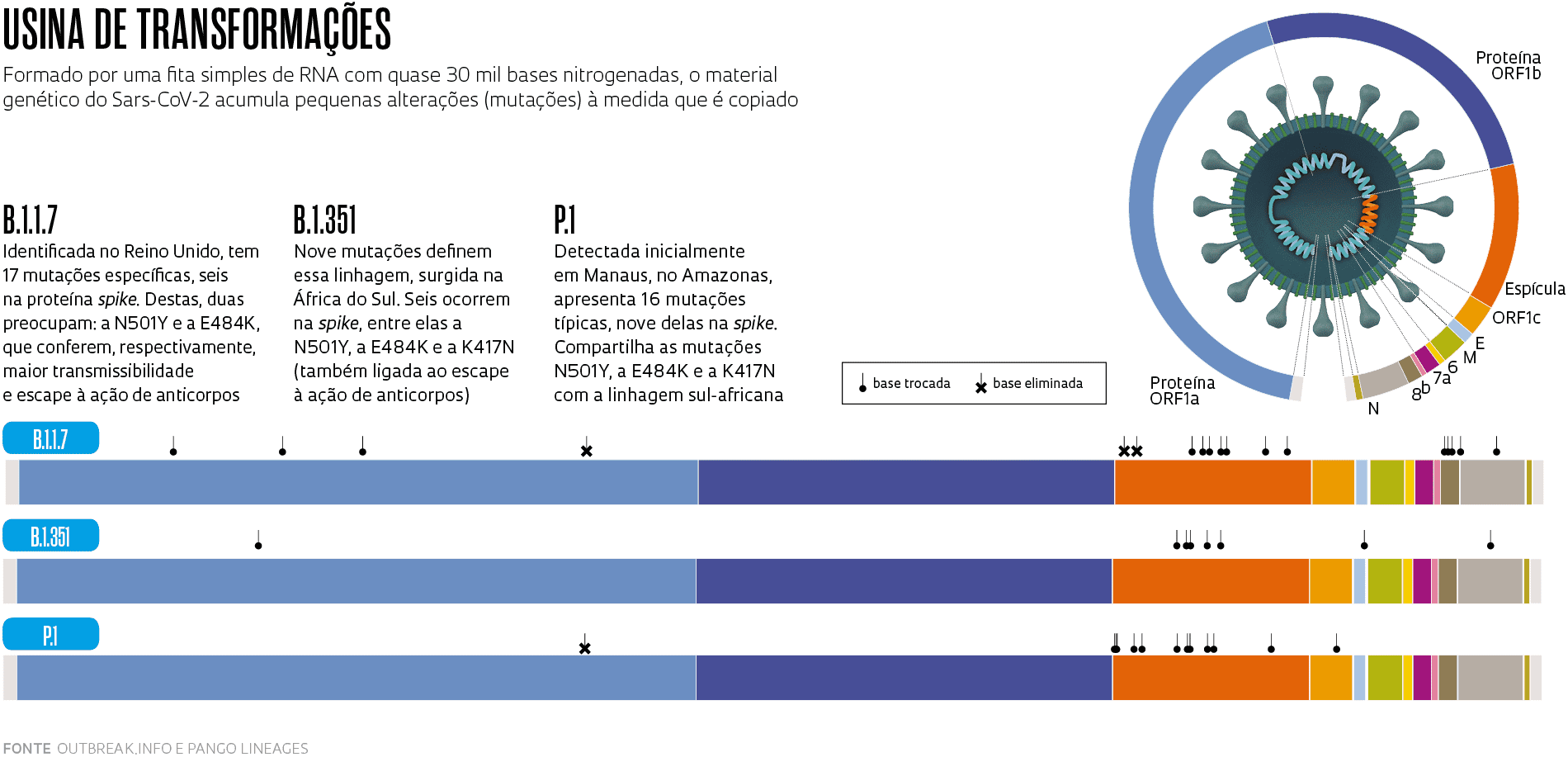
A primeira dessas variantes, que já se tornou uma linhagem e em março estava presente em 118 países, foi detectada em 14 de dezembro do ano passado no Reino Unido. Ela começou a circular em setembro no condado de Kent, no sudeste da Inglaterra, e rapidamente se espalhou. Apelidada inicialmente de variante de Kent ou britânica, tornou-se depois conhecida por um frio e sóbrio conjunto de letras e números (B.1.1.7) definido por uma nomenclatura proposta por pesquisadores da Austrália e do Reino Unido. Essa sequência de números indica que ela é a sétima variante derivada da primeira que descende da linhagem B.1, uma das duas que surgiu originalmente em Wuhan – a outra, possivelmente mais antiga, é a A.1, que desapareceu em meados do ano passado. Por causa de algumas alterações (mutações) que apresenta no genoma, a B.1.1.7 é transmitida ao menos duas vezes mais facilmente do que a linhagem que a originou e, ao que parece, também causa doença mais grave. No final de 2020 havia sinais de que ela poderia contribuir para o aumento das hospitalizações no Reino Unido e, agora, surgiram evidências de que está associada a um risco maior de morrer.
Em um estudo publicado em 15 de março na revista Nature, o epidemiologista Nicholas Davies e matemáticos e estatísticos da Escola de Higiene e Medicina Tropical de Londres estimaram que uma pessoa infectada por essa variante tem, em média, uma probabilidade 61% maior de morrer do que alguém que contraiu alguma das linhagens que circulavam anteriormente. Eles chegaram a essa conclusão depois de analisar 4.945 óbitos ocorridos em um grupo de 1.146.534 britânicos que haviam testado positivo para o novo coronavírus. Dias antes, pesquisadores liderados pelo médico Robert Challen, da Universidade de Exeter, também no Reino Unido, apresentaram um resultado semelhante em artigo publicado em 10 de março na revista The BMJ. Ao comparar o total de mortes em um grupo de 54.906 britânicos infectados com a B.1.1.7 com os óbitos em número igual de pessoas que haviam contraído outra linhagem do vírus, verificaram que o primeiro grupo tinha um risco de morrer 64% maior do que o segundo.
Apesar de se disseminar facilmente na população humana, o Sars-CoV-2, na fase inicial da pandemia, parecia ser um vírus razoavelmente bem-comportado, que sofria modificações muito lentamente. A cada mês acumulava, em média, duas mutações na sequência de quase 30 mil bases nitrogenadas, as letras químicas que compõem seu genoma, uma espécie de manual de instruções para a fabricação de novas cópias do vírus. Só para se ter um parâmetro de comparação, o vírus da influenza A, causador de pandemias de gripe, evolui muito mais rapidamente: sofre uma mutação a cada vez que se multiplica, em questão de horas, ritmo que obriga a atualização anual da composição da vacina contra a gripe.
Já no final de 2020, quando eram anunciados os resultados de eficácia das primeiras vacinas e ressurgia a esperança de que a pandemia estivesse se atenuando, o novo coronavírus voltou a surpreender. “Passaram a surgir variantes que apresentavam simultaneamente várias mutações e se disseminaram rapidamente, substituindo as anteriores”, lembra o virologista Fernando Spilki, pesquisador da Universidade Feevale, no Rio Grande do Sul, e coordenador da Rede Nacional de ômicas de Covid-19, a Corona-ômica BR, que acompanha a circulação do vírus no país.
Uma dessas variantes é a que originou a linhagem B.1.1.7. Ela apresenta 23 mutações em relação à B.1, de Wuhan, das quais 17 provocam a troca de um aminoácido nas proteínas do vírus – as outras seis são inócuas (ver infográfico abaixo). Os aminoácidos são compostos químicos que, unidos uns aos outros em sequência, formam as proteínas. Em alguns casos, a substituição de um único aminoácido é suficiente para alterar a estrutura tridimensional da proteína e modificar seu funcionamento.
Pesquisadores e profissionais da área da saúde se preocupam porque oito das substituições da B.1.1.7 (seis trocas e duas eliminações) ocorrem em alguns dos 1.273 aminoácidos da proteína spike (S), justamente a que permite ao vírus aderir à superfície das células humanas e invadi-las e é o principal alvo dos anticorpos produzidos por algumas vacinas. Uma mutação em especial chama a atenção: a N501Y. Essa mutação leva à substituição do aminoácido asparagina (N) pelo aminoácido tirosina (Y) na posição 501 da proteína S. Identificada pela primeira vez na linhagem B.1.1.7, essa troca parece aumentar a adesão do vírus às células e sua transmissibilidade. Por causa dessas características, a agência de saúde Public Health England (PHE), do Reino Unido, qualificou essa linhagem como variante de preocupação número 1 de dezembro de 2020.
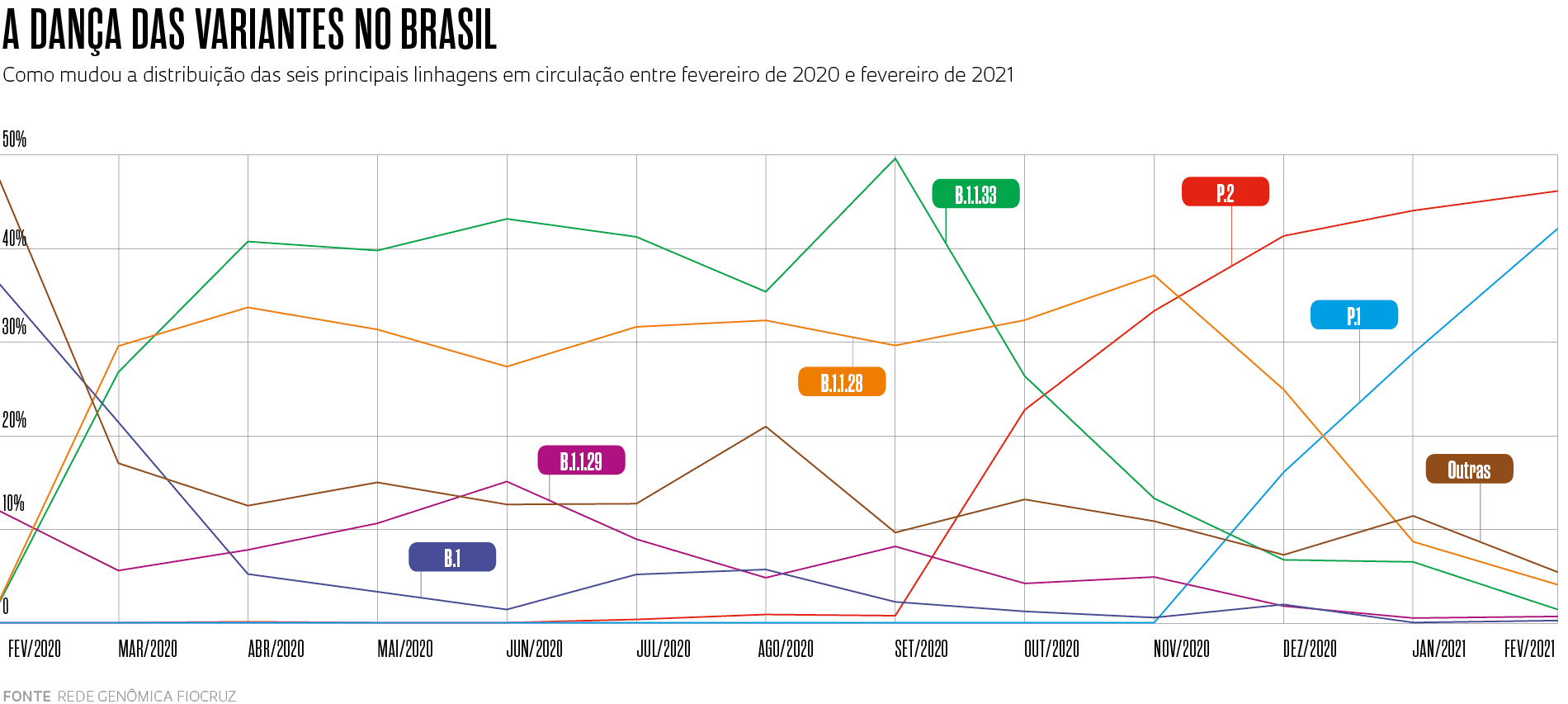
Em meados de março, segundo estimativa da Nextstrain baseada em dados do sistema Pangolin, a linhagem B.1.1.7 causava 42% das infecções pelo novo coronavírus no mundo (ver gráfico abaixo). De maneira independente e quase simultânea, a mutação N501Y também apareceu nas duas outras linhagens do vírus que mais inquietam os especialistas: a B.1.351, originária da África do Sul, e a P.1, que surgiu em Manaus, no Brasil. Elas se encontram em expansão e, estima-se, já causam, respectivamente, 6% e 2% dos casos de Covid-19 no planeta. “Até o surgimento da variante B.1.1.7 no Reino Unido, acreditávamos que a taxa de evolução do vírus fosse lenta e que seria possível segurar o aumento de sua diversidade até a chegada das vacinas”, conta a médica Ester Sabino, da Universidade de São Paulo (USP), coordenadora de uma das equipes que identificaram a P.1.
A linhagem B.1.351 foi detectada inicialmente na região metropolitana de Nelson Mandela Bay, no sul da África do Sul, em outubro de 2020, embora possivelmente tenha surgido meses antes, e foi reportada pelas autoridades do país em dezembro. Descrita pelo grupo coordenado pelo virologista brasileiro Túlio de Oliveira, da Universidade KwaZulu-Natal, em um artigo publicado em março deste ano na revista Nature, ela apareceu com mais frequência em pessoas mais jovens e saudáveis e hoje já tem transmissão local na Europa e na América do Norte.
Os vírus dessa linhagem também exibem 23 modificações em seu genoma, oito delas no gene da proteína S. Além da N501Y, duas outras mutações na spike deixam em alerta os especialistas: a E484K, que representa a troca de um glutamato (E) por uma lisina (K) e em fevereiro deste ano começou a ser detectada também na linhagem britânica; e a K417N, na qual uma lisina é reposta por uma asparagina (N). Essas substituições alteram uma região da spike chamada domínio de ligação ao receptor, que entra em contato mais intimamente com o receptor ACE2 na superfície das células humanas e abre caminho para a entrada do vírus, e impedem que certos anticorpos produzidos após a aplicação de algumas vacinas ou infecções prévias por outras variedades do Sars-CoV-2 se conectem ao vírus e o neutralizem. Estudos preliminares indicam que as pessoas infectadas pela B.1.351 – a segunda variante de preocupação detectada em dezembro de 2020, segundo o PHE – apresentam maior quantidade de vírus no organismo, o que pode facilitar a transmissão, além de potencialmente se beneficiarem menos dos efeitos protetores de algumas vacinas.
Essas três alterações na spike também estão presentes na linhagem P.1, originária do Brasil, que tem ainda outras sete alterações na mesma proteína. Ela descende da linhagem B.1.1.28, encontrada no país desde o início da pandemia, e foi nomeada com outra letra porque o sistema só aceita três conjuntos de algarismos. A linhagem brasileira possivelmente começou a circular em Manaus no início de novembro e foi detectada no mês seguinte, simultaneamente por pesquisadores da USP e da Fundação Oswaldo Cruz (Fiocruz), durante o aumento no número de internações que marcou o início da segunda onda da pandemia na região Norte do país, quando o sistema de saúde do Amazonas colapsou e pessoas morreram por asfixia em consequência da falta de oxigênio medicinal nos hospitais.
Um indivíduo infectado pela P.1 produz, em média, duas vezes mais vírus do que os contaminados pelas linhagens que circulavam antes no país, constatou a equipe do virologista Renato Santana de Aguiar, da Universidade Federal de Minas Gerais (UFMG), integrante da rede Corona-ômica BR. A presença de mais vírus no organismo aumenta de 1,4 a 2,2 vezes a possibilidade de transmissão, segundo estimativa calculada pelo grupo liderado por Sabino e pelo biomédico português Nuno Faria, da Universidade de Oxford, no Reino Unido. Outra equipe da USP, coordenada pelo biólogo Paulo Inácio Prado, verificou ainda que a P.1 tem uma probabilidade – baixa, é verdade, de 6,4% – de infectar novamente quem já foi contaminado por outras linhagens do vírus.
Alguns especialistas defendem que, diante dessas características da P.1, medidas mais drásticas de saúde pública deveriam ter sido adotadas no início do ano para tentar impedir a disseminação dessa linhagem, como um rígido controle de circulação de pessoas e o bloqueio de voos do Amazonas para outras regiões. Desde dezembro, no entanto, ao menos 120 mil pessoas deixaram o Amazonas rumo a outros estados e países – e até pacientes com Covid-19 foram transferidos para outras regiões brasileiras por falta de leitos.

Christopher Furlong / Getty images
Moradores de Manchester, no Reino Unido, fazem testagem em fevereiro deste ano, após surto com a B.1.1.7
Christopher Furlong / Getty imagesUma das consequências é que, em meados de março, a P.1 já representava 41% das infecções pelo novo coronavírus no Brasil (ver gráfico). Outra análise realizada pela Rede Genômica da Fiocruz indicava que em menos de três meses a linhagem se tornou responsável por mais da metade das infecções em seis estados: Pernambuco (51%), Rio Grande do Sul (63%), Rio de Janeiro (63%), Santa Catarina (64%), Paraná (70%) e Ceará (71%).“A situação que o país vive hoje é semelhante àquela em que se encontrava Manaus em dezembro, com a P.1 em franca disseminação”, afirma Sabino, que coordena no país o Centro Conjunto Brasil-Reino Unido para Descoberta, Diagnóstico, Genômica e Epidemiologia de Arbovírus (Cadde), financiado pela agência britânica Medical Research Council e pela FAPESP.
“O Brasil tornou-se um terreno fértil para o surgimento de variantes e linhagens mais contagiosas”, afirma o virologista Eurico Arruda, da USP em Ribeirão Preto, que estuda os coronavírus há quase três décadas (ver Pesquisa FAPESP nº 301). A população suscetível a contrair o vírus é grande, o país vacina muito lentamente e as medidas de prevenção, como distanciamento social e uso de máscara, não vêm sendo adotadas de forma efetiva em todas as regiões. “Quanto mais gente o vírus infectar, mais ele vai se multiplicar e sofrer alterações”, explica.
Vírus são seres naturalmente propensos a acumular mutações em seu genoma porque, diferentemente dos seres vivos formados por células, não têm mecanismos eficientes de correção de erros. À medida que o material genético viral é copiado para gerar novos exemplares, alterações ocorrem aleatoriamente, de modo semelhante ao que acontece com alguém que copia à mão um texto longo (mesmo que de modo muito atento). Sem proteínas que detectem com eficácia as incorreções e as desfaçam, as mutações acabam incorporadas ao material genético dos novos vírus – no caso do Sars-CoV-2, uma molécula de RNA de fita simples.
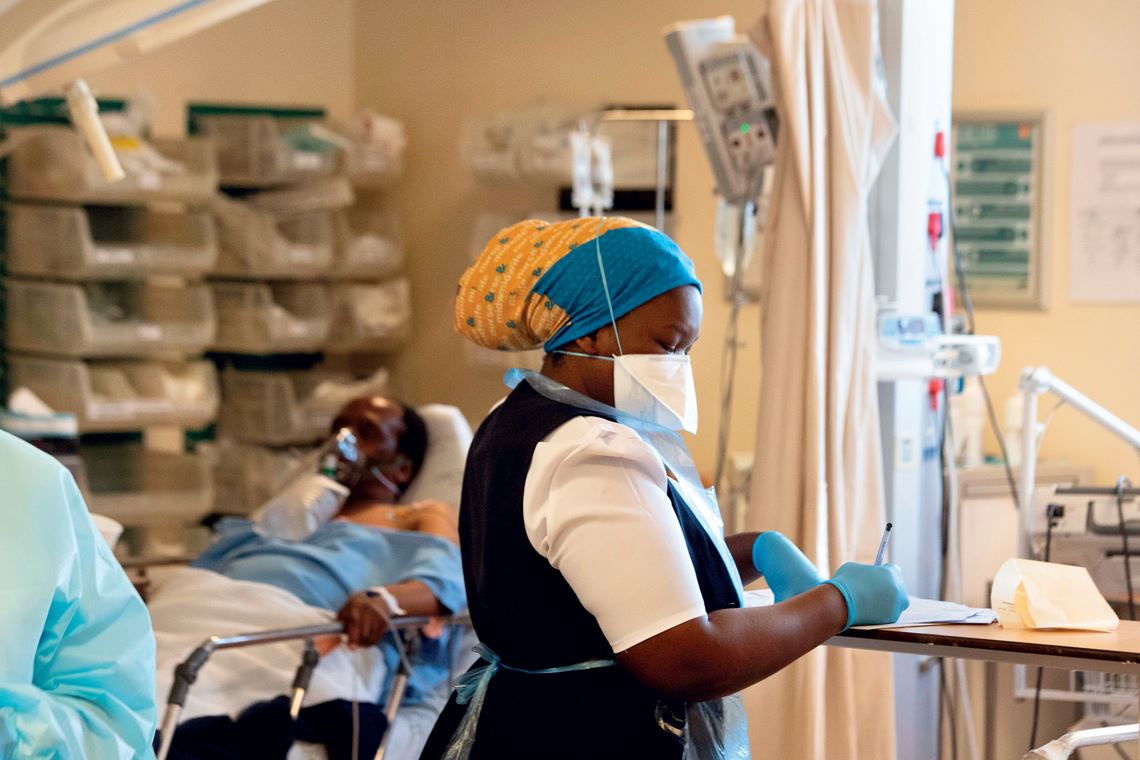
Rodger Bosch / AFP via Getty images
Profissional da saúde acompanha paciente com Covid-19 na África do Sul, após o surgimento de uma nova variante
Rodger Bosch / AFP via Getty images“Mutações ocorrem com frequência, mas nem todas são importantes para aumentar a transmissibilidade ou a patogenicidade do vírus”, explica a bioquímica Marilda Siqueira, do Laboratório de Vírus Respiratórios e do Sarampo, da Fiocruz, referência em vigilância genômica do coronavírus para o Ministério da Saúde e para a OMS. Outras, no entanto, podem conferir vantagem adaptativa ao vírus e permitir, por exemplo, que penetre mais facilmente nas células, multiplique-se mais ou escape à ação dos anticorpos. Nessas situações, elas se fixam no genoma do vírus e são transmitidas para as gerações futuras.
“Suspeitamos que esse seja o caso das mutações N501Y, E484K e K417N, que apareceram de modo independente em variantes surgidas em diferentes lugares do mundo e parecem aumentar a aptidão viral”, conta Aguiar, da UFMG. Além de estar presente na P.1, na B.1.351 de origem sul-africana e desde o início do ano também na britânica B.1.1.7, ela também aparece no material genético de duas outras variantes brasileiras, identificadas com auxílio do grupo de Siqueira: a P.2, que já se distribui por todo o país, e a N.9, detectada no início de março. Elas ainda estão sob investigação e por enquanto não preocupam como a P.1.
“Minha expectativa é que, com o tempo, surjam variantes mais transmissíveis, mas que causem uma doença mais branda, como ocorreu com o vírus da influenza A, que causou a pandemia de gripe espanhola em 1918”, diz Arruda. Publicada em 9 de março na revista Science, uma análise da diversidade genética do Sars-CoV-2 em 1.313 pessoas feita pela epidemiologista Katrina Lythgoe, da Universidade de Oxford, e colaboradores sugere que o desenvolvimento de variantes mais transmissíveis e que escapem à ação do sistema de defesa seja incomum. Uma vez que emergem, no entanto, elas podem se disseminar rapidamente.
Além de permitir ao vírus se espalhar com mais facilidade, as mutações encontradas nas novas linhagens vêm tirando o sono de muitos especialistas pela ameaça que podem representar para a eficácia das vacinas. Os imunizantes produzidos pelas farmacêuticas norte-americanas Janssen e Novavax, por exemplo, já mostraram ser menos eficientes contra a B.1.351, da África do Sul. Assim como a Novavax, a vacina desenvolvida pela Universidade de Oxford com a farmacêutica anglo-sueca AstraZeneca também perde um pouco do seu efeito ante a linhagem B.1.1.7, do Reino Unido. Seu desempenho, porém, é muito pior contra a linhagem da África do Sul. Ela mostrou uma importante redução na capacidade de evitar os casos leves e moderados de doença causada pela B.1.351 (não houve casos graves no grupo vacinado nem no que recebeu placebo), de acordo com os resultados de um ensaio clínico com cerca de 2 mil participantes publicados em 16 de março na revista New England Journal of Medicine.
Estudos realizados pelo grupo de Módena, na Unicamp, e pela equipe do imunologista Michael Diamond, da Universidade de Washington em Saint Louis, Estados Unidos, identificaram, respectivamente, algum nível de perda de efeito de anticorpos produzidos pela CoronaVac e pela vacina da Pfizer contra a linhagem P.1 em testes in vitro. Ainda não é possível, no entanto, afirmar que essas variantes escapem do efeito das vacinas em pessoas. A literatura científica sobre o tema ainda traz contradições e outros trabalhos chegaram a conclusões diferentes. Mesmo assim, no final de fevereiro alguns fabricantes de vacinas anunciaram que estão trabalhando em doses de reforço e formulações que contemplem as mudanças das variantes.
Além disso, os anticorpos não funcionam apenas aderindo ao vírus e bloqueando sua entrada nas células. Eles podem desencadear reações químicas que são lesivas para as partículas virais ou servir de sinalizadores, marcando os vírus para que sejam engolfados e digeridos por células de defesa. “Avaliar outras estratégias do sistema imune, como a atividade das células de defesa, é mais laborioso, mas essencial para se chegar a conclusões mais robustas”, conta o biomédico William Souza, que faz estágio de pós-doutorado na Universidade de Oxford e colaborou nos experimentos de Módena e de Sabino. “As vacinas continuam evitando que as pessoas adoeçam e morram.”
Para Módena, os resultados obtidos até o momento contra as novas linhagens do vírus reforçam a ideia de que mesmo quem já teve a doença ou foi vacinado precisa manter os cuidados e continuar usando máscaras e adotando medidas de higiene e distanciamento social. “O que se sabe até o momento indica que quem já teve Covid-19 ou mesmo quem já recebeu algumas das vacinas pode ser infectado pelo vírus de uma das linhagens de preocupação”, afirma. Em um levantamento realizado em janeiro pela revista Nature com 100 virologistas, imunologistas e especialistas em doenças infecciosas de diferentes países, 90% afirmaram imaginar que o vírus pode se tornar endêmico e continuar circulando em pequenos bolsões nos próximos anos – seja porque escapa à ação dos anticorpos, porque não haverá vacinas suficientes para todos, porque alguns se recusam a ser vacinados ou porque eles continuaram a infectar animais na natureza.
O nascimento das linhagens Sequenciamento de genoma permite acompanhar as transformações do vírusPara acompanhar o surgimento das variantes e linhagens, os especialistas sequenciam o material genético dos vírus em circulação em certo momento e determinada região e comparam com o das que existiam previamente. Até o início de março, haviam sido sequenciados no mundo quase 670 mil genomas do Sars-CoV-2 – cerca de 4 mil sequenciamentos foram feitos no Brasil. O país conta hoje com ao menos três redes de laboratórios que fazem a vigilância genômica de forma sistemática: a Rede Genômica da Fiocruz, financiada pelo Ministério da Saúde; a Rede Nacional de ômicas de Covid-19 (Corona-ômica BR), apoiada pelo Ministério de Ciência, Tecnologia e Inovações; e o Centro Conjunto Brasil-Reino Unido para Descoberta, Diagnóstico, Genômica e Epidemiologia de Arbovírus (Cadde), custeado pela agência britânica Medical Research Council e pela FAPESP. “Precisamos ampliar nossa capacidade de sequenciamento e estamos iniciando esse projeto”, conta a bioquímica Marilda Siqueira, pesquisadora da Rede Genômica da Fiocruz. “Mesmo assim, fomos capazes de identificar a introdução da linhagem britânica em vários estados e de detectar o início da circulação da P.1, da P.2 e da N.9.” O sequenciamento do genoma depende do uso de reagentes importados, leva dias e custa cerca de R$ 600. Para melhorar a vigilância, alguns laboratórios nacionais desenvolveram testes moleculares (PCR) que permitem detectar as novas linhagens do vírus. Um deles é o da Fiocruz no Amazonas. Outro é o do virologista Renato Santana Aguiar, da UFMG, que identifica a P.1, a P.2, a B.1.1.7 e a B.1.351. “O teste sai por R$ 60 e fica pronto em horas”, conta Aguiar. “Ele permitirá conhecer com mais detalhes a circulação dessas linhagens.” A genotipagem por PCR, no entanto, não substitui o sequenciamento, única forma de identificar o surgimento de novas variantes e linhagens.
Dando nome ao vírus Sistemas adotam estratégias distintas para denominar linhagensQuem já leu algo sobre as novas linhagens e variantes do novo coronavírus se deparou com sequências de letras e números que parecem não fazer sentido. São tentativas de organizar o conhecimento sobre o vírus e acompanhar sua dispersão e evolução. Nos últimos meses, ao menos três estratégias de nomeá-los se tornaram conhecidas. Uma delas é um sistema proposto pelo biólogo Andrew Rambaut, da Universidade de Edimburgo, na Escócia. Conhecido pela sigla Pangolin, acrônimo de Phylogenetic Assignment of Named Global Outbreak Lineages, ele fornece informações mais detalhadas sobre a circulação do vírus em determinado momento. Nesse sistema, as variantes ou linhagens são classificadas pelo grau de parentesco evolutivo, atribuindo uma letra seguida de até três algarismos. Nele, a linhagem surgida no Reino Unido é a B.1.1.7, a sétima variante derivada da primeira que descende da linhagem B.1, uma das duas que apareceram originalmente em Wuhan. A linhagem sul-africana é a B.1.351 (351ª descendente da B.1). Já a brasileira, por ser descendente da linhagem B.1.1.28, que já tinha três números, recebeu uma nova letra e se tornou a P.1. Um segundo sistema de classificação é o Nextstrain, proposto pelo grupo de Trevor Bedford, do Centro de Pesquisa para o Câncer Fred Hutchinson, nos Estados Unidos, e colaboradores. Ele agrupa os vírus em conjuntos maiores (clados), que são descritos pelo ano de identificação do clado, seguido de uma letra e do nome de uma mutação definidora do grupo. Assim, a linhagem do Reino Unido integrou o clado 20I/501Y.V1; a da África do Sul o 20H/501.V2 e do Brasil o 20J/501.V3. O terceiro sistema de classificação é de um consórcio internacional criado em 2008, o Gisaid. Adotado pela OMS, também reúne os vírus por clados, definidos por uma ou mais letras que integram as principais mutações do grupo. Nela, a linhagem do Reino Unido está no clado GRY, a da África do Sul no GH e a do Brasil no GR.
Projetos
1. Centro conjunto Brasil-Reino Unido para descoberta, diagnóstico, genômica e epidemiologia de arbovírus (Cadde) (nº 18/14389-0); Modalidade Projeto Temático; Pesquisadora responsável Ester Cerdeira Sabino (USP); Investimento R$ 5.331.725,16.
2. ICTP – Instituto sul-americano para física fundamental: Um centro regional para física teórica (nº 16/01343-7); Modalidade Projeto Temático; Pesquisador responsável Nathan Jacob Berkovits (Unesp); Investimento R$ 15.421.379,38.
3. Patogênese e neurovirulência de vírus emergentes no Brasil (nº 16/00194-8); Modalidade Jovem Pesquisador; Pesquisador responsável José Luiz Proença Módena (Unicamp); Investimento R$ 2.509.395,80.
4. Caracterização de fatores de risco intrínsecos e o desenvolvimento de novas alternativas de diagnóstico e tratamento para Covid-19 (nº 20/04558-0); Modalidade Auxílio à Pesquisa – Regular; Pesquisador responsável José Luiz Proença Módena (Unicamp); Investimento R$ 184.077,00.
5. Replicação e efeitos celulares de rinovírus em tecidos linfoides (nº 18/25605-6); Modalidade Auxílio à Pesquisa – Regular; Pesquisador responsável Eurico de Arruda Neto (FMRP-USP); Investimento R$ 236.000,00.
Artigos científicos
DAVIES, M. G. et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. 15 mar. 2021.
CHALLEN, R. et al. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. The BMJ. 10 mar. 2021.
TEGALLY, H. et al. Emergence of a SARS-CoV-2 variant of concern with mutations in spike glycoprotein. Nature. 9 mar. 2021.
NAVECA, F. et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein. Virological.org. 11 jan. 2021.
FARIA, N. R. et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. Virological.org. 12 jan. 2021
FUJINO, T. et al. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan. Emerging Infectious Diseases. 10 fev. 2021.
NAVECA, F. et al. COVID-19 epidemic in the Brazilian state of Amazonas was driven by long-term persistence of endemic SARS-CoV-2 lineages and the recent emergence of the new Variant of Concern P.1. Research Square. 25 fev. 2021.
RESENDE, P. C. et al. A potential SARS-CoV-2 variant of interest (VOI) harboring mutation E484K in the Spike protein was identified within lineage B.1.1.33 circulating in Brazil. medRxiv. 13 mar. 2021.
FARIA, N. R. et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv. 3 mar. 2021.
COUTINHO, R. M. et al. Model-based estimation of transmissibility and reinfection of Sars-CoV-2 P.1 variant. medRxiv. 9 mar. 2021.
LYTHGOE, K. A. et al. Sars-CoV02 within-host diversity and transmission. Science. 9 mar. 2021.
MADHI, S. A. et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. New England Journal of Medicine. 16 mar. 2021.
SOUZA, W. M. et al. Levels of Sars-CoV-2 lineage P.1 neutralization by antibodies elicited after natural infection and vaccination. Pre-prints with The Lancet. 1º mar. 2021.
CHEN, R. E. et al. Resistance of Sars-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nature Medicine. 4 mar. 2021.
LIU, Y. et al. Neutralizing Activity of BNT162b2-Elicited Serum. New England Journal of Medicine. 8 mar. 2021.
Republicar