In the year since the pandemic was officially declared by the World Health Organization (WHO) on March 11, 2020, the world has watched in awe as 122 million people have fallen ill and at least 2.7 million have died—a third of these deaths occurred in just three countries: the USA, Brazil, and Mexico. The public has been able to follow the evolution of the pathogen almost in real time and with an unprecedented level of detail, watching as it brought health systems in many countries to the brink of collapse and profoundly affected economies. Since it was first identified in the city of Wuhan, China, in late 2019, the SARS-CoV-2 virus has undergone a series of transformations as it spreads across the planet. As it infects more and more people and continuously replicates, it has accumulated small changes in its genetic code until, in different times and places, it has become so different from the original that it is classified as a new variant, which after prospering and spreading, becomes known as a lineage. Lineages include specimens of the same origin that are very similar, but may have small differences.
On March 15 this year, Nextstrain, a tool for visualizing viral genomes, listed no less than 359 lineages of the novel coronavirus cataloged by the PANGOLIN classification system since December 2019 (see page 24). Of this high number, there are three—one from the United Kingdom, another from South Africa, and the third from Brazil—that have been causing particular concern because they are more transmissible, may be resistant to antibodies, and in some cases, cause more serious cases of the disease than previous strains. “With the genetic tools currently available to us, we are watching the pathogen’s evolution in real time,” says virologist José Luiz Proença Módena, head of the Laboratory for Studies of Emerging Viruses at the University of Campinas (UNICAMP).
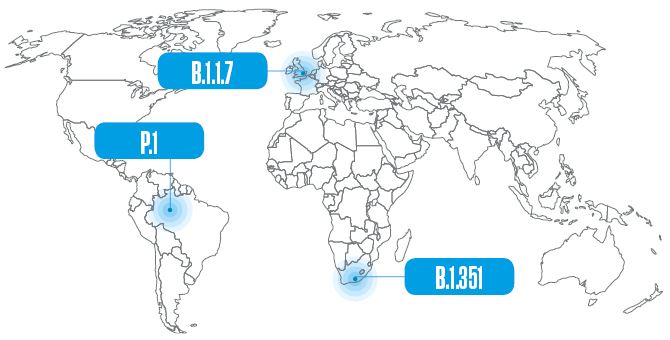
The first of these variants, which has already become a lineage and had been identified in 118 countries by March, was first detected on December 14 last year in the UK. It began circulating in September in the county of Kent, in southeast England, and quickly spread nationwide. Initially called the Kent or British variant, it later became known by a more standardized set of letters and numbers (B.1.1.7), according to a nomenclature proposed by researchers from Australia and the UK. This sequence indicates that it is the seventh variant derived from the first that descended from the B.1 lineage, one of the two that originally appeared in Wuhan. The other, possibly older lineage is A.1, which disappeared in the middle of last year. Due to some of the alterations (mutations) to its genome, B.1.1.7 spreads twice as fast as the lineage it originated from, and seems to cause more serious cases of the disease. At the end of 2020 there were signs that it may be contributing to the increased number of hospitalizations in the UK, and now there is evidence that it is associated with higher fatality.
In a study published in the journal Nature on March 15, epidemiologist Nicholas Davies and mathematicians and statisticians from the London School of Hygiene & Tropical Medicine estimated that a person infected with this variant is on average 61% more likely to die than someone who contracted one of the previous strains. The conclusion was reached after analyzing 4,945 deaths from a group of 1,146,534 Britons who had tested positive for the novel coronavirus. Days earlier, a group of researchers led by physician Robert Challen from the University of Exeter, UK, presented a similar result in an article published in the journal The BMJ on March 10. After comparing the total number of deaths from a group of 54,906 Britons infected with B.1.1.7 to the number of deaths in a group of equal size who had contracted other strains of the virus, they found that the former were 64% more likely to die.
Despite spreading easily through the human population, in the early stages of the pandemic SARS-CoV-2 appeared to be a reasonably stable virus that mutated relatively slowly. Every month, it accumulated an average of two mutations to its sequence of almost 30,000 nitrogenous bases, the chemical elements that compose its genome, a kind of instruction manual for manufacturing new copies of the virus. By way of comparison, the influenza A virus, which causes flu pandemics, evolves much faster: it mutates every time it multiplies, in a matter of hours, which is why the vaccine has to be updated every year.
At the end of 2020, when the effectiveness of the first vaccines were announced and people began to hope that the pandemic was subsiding, the novel coronavirus provided a new surprise. “Variants started to appear with several mutations and spread quickly, replacing previous strains,” says virologist Fernando Spilki, a researcher at Feevale University in the state of Rio Grande do Sul and head of the Brazilian National COVID-19 Omics Network (Corona-ômica BR), which tracks the virus’s circulation in the country.
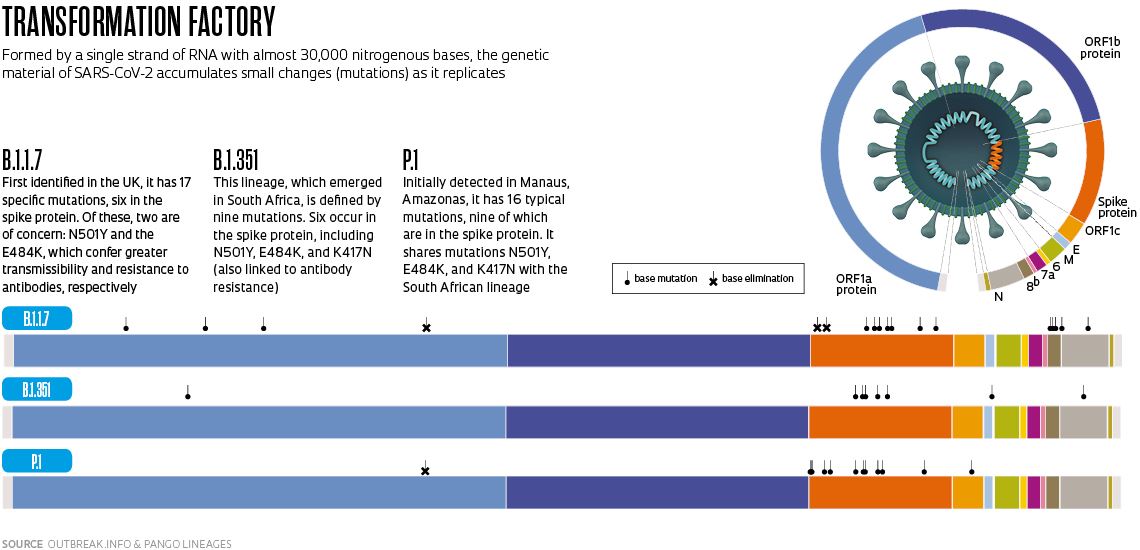
It was one of these variants that gave rise to the B.1.1.7 lineage. It has 23 mutations from the Wuhan B.1 strain, of which 17 alter an amino acid in the virus’s proteins—the other six are innocuous (see infographic below). Amino acids are chemical compounds that join together in sequence to form proteins. In some cases, altering a single amino acid is enough to change the protein’s three-dimensional structure and modify how it functions.
The B.1.351 lineage was initially detected in the Nelson Mandela Bay Metropolitan Municipality in South Africa in October 2020—although it may actually have appeared months earlier—and was reported by the country’s authorities in December. Described by a group led by Brazilian virologist Túlio de Oliveira of the University of KwaZulu-Natal in an article published in the journal Nature in March this year, it was seen more frequently in younger, healthier people and has now been transmitted locally in Europe and North America.
The viruses of this lineage also have 23 genome modifications, eight of which are on the spike protein. In addition to N501Y, two other mutations on the spike have experts on alert: E484K, in which a glutamate (E) is replaced with a lysine (K), a mutation that was also detected also in the British variant in February of this year; and K417N, in which a lysine is replaced by an asparagine (N). These substitutions alter a region of the spike protein called the receptor-binding domain, which comes into close contact with the ACE2 receptor on the surface of human cells and opens the way for the virus to enter, and prevents certain antibodies produced as a result of some vaccines or previous infections by other SARS-CoV-2 variants from connecting to the virus and neutralizing it. Preliminary studies indicate that people infected with B.1.351—the second variant of concern detected in December 2020, according to PHE—have a greater viral load in their bodies, which can increase transmission and potentially reduce the protective effects of some vaccines.
These three changes to the spike protein are also present in the P.1 strain, originally from Brazil, which also has seven other alterations in the same protein. It descends from the B.1.1.28 lineage, which has been circulating in the country since the beginning of the pandemic, and was given a new letter because the coding system only accepts three sets of numbers. The Brazilian lineage may have begun circulating in Manaus in early November and was detected in the following month, simultaneously by researchers from USP and the Oswaldo Cruz Foundation (FIOCRUZ), as the number of hospitalizations increased in the region, marking the start of the pandemic’s second wave in the northern Brazil, when the health system in Amazonas collapsed and people died of asphyxiation due to a lack of medicinal oxygen in hospitals.
An individual infected with the P.1 strain has twice the viral load on average as someone contaminated by the previous strains found in the country, according to a team led by virologist Renato Santana de Aguiar of the Federal University of Minas Gerais (UFMG), a member of the Corona-ômica BR network. A research group led by Sabino and Portuguese biomedical expert Nuno Faria of the University of Oxford, UK, calculated that a higher viral load makes the chance of transmission between 1.4 and 2.2 times more likely. Another team from USP, led by biologist Paulo Inácio Prado, found that P.1 can infect people who have already had other strains of the virus—although the probability is low, at 6.4%.
Some experts argue that based on these characteristics, stricter public health measures should have been adopted at the beginning of the year to try to prevent the spread of the lineage, such as restrictions on movement and suspending flights from Amazonas to other regions. Since December, however, at least 120,000 people have left Amazonas to visit other states and countries—and even COVID-19 patients have been transferred from the area to other Brazilian regions due to a lack of hospital beds.
One consequence is that in mid-March, the P.1 strain already accounted for 41% of novel coronavirus infections in Brazil (see graph on page 18). Another analysis by FIOCRUZ’s Genome Network indicated that in less than three months, the strain became responsible for more than half of all infections in six states: Pernambuco (51%), Rio Grande do Sul (63%), Rio de Janeiro (63%), Santa Catarina (64%), Paraná (70%), and Ceará (71%). “The situation the country is facing now is similar to what Manaus went through in December, with P.1 spreading widely,” says Sabino, who is Brazil’s head of the The Brazil-UK Center for Arbovirus Discovery, Diagnosis, Genomics, and Epidemiology (CADDE), funded by the British Medical Research Council and FAPESP.
“Brazil has become a breeding ground for more contagious variants and strains,” says virologist Eurico Arruda of USP in Ribeirão Preto, who has been studying coronaviruses for almost three decades (see Pesquisa FAPESP issue no. 301). The population susceptible to contracting the virus is large, Brazil’s immunization program is very slow, and preventive measures, such as social distancing and use of masks, have not been widely adopted in all regions. “The more people the virus infects, the more it will multiply and mutate,” he explains.
Viruses naturally accumulate mutations in their genome because unlike living beings formed by cells, they do not have efficient error correction mechanisms. As the viral genetic material is replicated to generate new copies, alterations occur at random, similar to what would happen if a person were to copy a long piece of text by hand (even if they were doing so very attentively). Without proteins that effectively detect and correct genetic mistakes, these mutations are incorporated into the genetic material of new copies of the virus—which in the case of SARS-CoV-2, is a single-stranded RNA molecule.
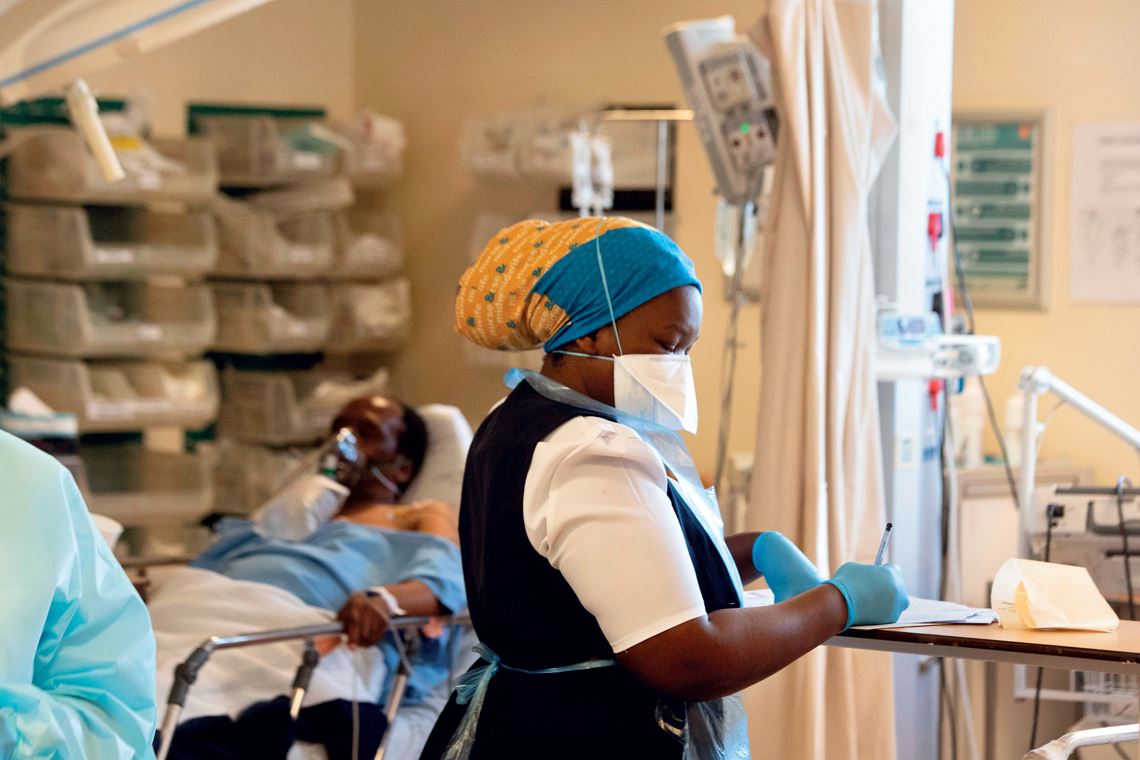
Rodger Bosch / AFP via Getty Images
A healthcare professional monitors a COVID-19 patient in South Africa, after the emergence of a new variant
Rodger Bosch / AFP via Getty Images“Mutations occur frequently, but not all have an effect on the transmissibility or pathogenicity of the virus,” explains biochemist Marilda Siqueira, from FIOCRUZ’s Laboratory of Measles and Respiratory Viruses, renowned for its work on the coronavirus’s genome for the Brazilian Ministry of Health and the WHO. Some, however, can give the virus an adaptive advantage, allowing it to infiltrate cells more easily, multiply faster, or resist antibodies, for example. In these situations, the virus is more successful and the genome changes are passed onto future generations.
“We suspect that this is the case with the N501Y, E484K, and K417N mutations, which appeared independently in variants that emerged in different places around the world and seem to increase viral capability,” says Aguiar, from UFMG. As well as being present in the P.1 lineage, in B.1.351 from South Africa, and also in the British lineage B.1.1.7 since the beginning of the year, the mutation also appears in the genetic material of two other Brazilian variants, identified with the help of Siqueira’s group: P.2, which has already spread nationwide, and N.9, detected in early March. They are still under investigation, but for now they are not as concerning as P.1.
“I expect that over time, more transmissible variants will emerge, but that they will cause a milder version of the disease, as occurred with the influenza A virus that caused the Spanish flu pandemic in 1918,” says Arruda. Published in the journal Science on March 9, an analysis of SARS-CoV-2’s genetic diversity in 1,313 people by epidemiologist Katrina Lythgoe of the University of Oxford and her colleagues suggests that more transmissible variants capable of evading the immune system are rare. Once they emerge, however, they can spread quickly.
As well as allowing the virus to spread more easily, the mutations found in the new strains have kept many experts awake at night because of the threat they may pose to vaccine effectiveness. The vaccines produced by American pharmaceutical companies Janssen and Novavax, for example, have already been shown to be less effective against the South African lineage B.1.351. Similarly, Novavax, the vaccine developed by the University of Oxford in partnership with Anglo-Swedish pharmaceutical company AstraZeneca, has less of an effect on B.1.1.7, the lineage that emerged in the UK. Its effectiveness against the South African lineage, however, is even worse. Its ability to prevent mild and moderate cases of disease caused by B.1.351 is significantly lower (there were no serious cases in the vaccinated group or in the placebo group), according to a clinical trial of about 2,000 participants published in the New England Journal of Medicine on March 16.
Studies carried out by Módena’s group at UNICAMP and a team led by immunologist Michael Diamond, from the University of Washington in Saint Louis, USA, identified a reduction in the effect of antibodies produced by CoronaVac and the Pfizer vaccine respectively against the P.1 strain during in vitro tests. It is not yet possible, however, to say that these variants are able to evade the vaccines. The scientific literature on the subject still demonstrates contradictions, with different papers reaching different conclusions. Even so, some vaccine manufacturers announced at the end of February that they are working on booster doses and formulations designed to work against new variants.
In addition, antibodies do not function only by binding to the virus and stopping it from infiltrating cells. They can also trigger chemical reactions that are harmful to viral particles or serve as flags, marking viruses to be engulfed and digested by immune cells. “Assessing other immune system strategies, such as the activity of immune cells, is more laborious, but essential to arrive at more robust conclusions,” says biomedical expert William Souza, who is doing a postdoctoral fellowship at the University of Oxford and participated in both Módena’s and Sabino’s experiments. “Vaccines continue to prevent people from getting sick and dying.”
For Módena, the results obtained so far against new strains of the virus reinforce the idea that even those who have already had the disease or been vaccinated need to continue wearing masks and following hygiene measures and social distancing. “What we know so far indicates that those who have had COVID-19 or even some of those who have already been vaccinated can be infected with one of the variants of concern,” he says. In a survey of 100 virologists, immunologists, and infectious disease specialists from around the world carried out by the journal Nature in January, 90% of respondents said they believed the virus could become endemic and continue to circulate in small pockets for years to come—either because it is resistant to antibodies, because there will not be enough vaccines for everyone, because some people refuse to be vaccinated, or because it continues to infect animals in the wild.
The birth of new lineages Genome sequencing used to track transformations of the virusTo monitor the emergence of variants and strains, experts sequence the genetic material of viruses in circulation at a given time and place, comparing them with those that previously circulated. By the beginning of March, almost 670,000 SARS-CoV-2 genomes had been sequenced worldwide—including about 4,000 in Brazil. The country currently has at least three laboratory networks performing systematic genome surveillance: FIOCRUZ’s Genome Network, funded by the Ministry of Health; the Brazilian National COVID-19 Omics Network (Corona-ômica BR), funded by the Ministry of Science, Technology, and Innovations; and the Brazil-UK Center for Arbovirus Discovery, Diagnosis, Genomics, and Epidemiology (CADDE), funded by the British Medical Research Council and FAPESP.
“We need to expand our sequencing capacity and we are starting to do so,” says biochemist Marilda Siqueira, a researcher from FIOCRUZ’s Genome Network. “We were able to identify the arrival of the British lineage in several Brazilian states and to detect the early circulation of P.1, P.2, and N.9.”
The genome sequencing process requires imported reagents, takes days to complete, and costs about R$600. To improve surveillance, some Brazilian laboratories have developed molecular tests (PCR) that can detect new strains of the virus. One is a FIOCRUZ lab in Amazonas. Another is the lab of virologist Renato Santana Aguiar, from UFMG, who identifies the P.1, P.2, B.1.1.7, and B.1.351 lineages. “The test costs R$60 and is ready in hours,” says Aguiar. “It allows us to learn about the circulation of these strains in more detail.” Genotyping by PCR, however, is not a substitute for sequencing, which is the only way to identify the emergence of new variants and strains.
Naming the virus Different systems used to name new strainsAnyone who has read anything about the new strains and variants of the novel coronavirus has come across sequences of letters and numbers that seem to make no sense. These codes are attempts to organize knowledge about the virus and monitor its spread and evolution. In the past few months, at least three naming strategies have come to light. One is a system proposed by biologist Andrew Rambaut, from the University of Edinburgh in Scotland. Known as PANGOLIN, which stands for Phylogenetic Assignment of Named Global Outbreak Lineages, it provides more detailed information about how the virus is circulating at any given time. In this system, variants or lineages are classified by their evolutionary distance from their predecessors, assigned a letter followed by up to three digits. The lineage that emerged in the UK, for example, is known as B.1.1.7, the seventh variant derived from the first that descends from lineage B.1, one of the two that originally appeared in Wuhan. The South African lineage is B.1.351 (the 351st descendant of B.1). The Brazilian strain, however, is a descendant of the B.1.1.28 line, which already had three numbers. It was thus given a new letter and became P.1. The second classification system is Nextstrain, proposed by a group led by Trevor Bedford, from the Fred Hutchinson Cancer Research Center, USA, and colleagues. It divides viruses into larger groups (known as clades), which are described by the year they were identified, followed by a letter and the name of one of the clade’s defining mutations. The UK lineage is therefore part of clade 20I/501Y.V1; South Africa’s is in 20H/501.V2, and Brazil’s is in 20J/501.V3. The third classification system was developed by GISAID, an international consortium established in 2008. Adopted by the WHO, it also groups viruses by clades, given one or more letters that include the group’s main mutations. In this system, the UK lineage is part of the GRY clade, South Africa’s is in the GH clade, and Brazil’s is in GR.
Projects
1. UK-Brazil Joint Centre for Arbovirus Discovery, Diagnosis, Genomics and Epidemiology (Cadde) (nº 18/14389-0); Grant Mechanism Thematic Project; Principal Investigator Ester Cerdeira Sabino (USP); Investment R$5,331,725.16.
2. ICTP – South American Institute for Fundamental Physics: A regional center for theoretical physics (nº 16/01343-7); Grant Mechanism Thematic Project; Principal Investigator Nathan Jacob Berkovits (UNESP); Investment R$15,421,379.38.
3. Pathogenesis and neurovirulence of emerging viruses in Brazil (nº 16/00194-8); Grant Mechanism Young Investigator Award; Supervisor José Luiz Proença Módena (UNICAMP); Investment R$2,509,395.80.
4. Characterization of intrinsic risk factors and the development of new alternatives for diagnose and treatment of Covid-19 (nº 20/04558-0); Grant Mechanism Regular Research Grant; Principal Investigator José Luiz Proença Módena (UNICAMP); Investment R$184,077.00.
5. Replication and cellular effects of rhinoviruses in lymphoid tissues (nº 18/25605-6); Grant Mechanism Regular Research Grant; Principal Investigator Eurico de Arruda Neto (FMRP-USP); Investment R$236,000.00.
Scientific articles
DAVIES, M. G. et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. Mar. 15, 2021.
CHALLEN, R. et al. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. The BMJ. Mar. 10, 2021.
TEGALLY, H. et al. Emergence of a SARS-CoV-2 variant of concern with mutations in spike glycoprotein. Nature. Mar. 9, 2021.
NAVECA, F. et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein. Virological.org. Jan. 11, 2021.
FARIA, N. R. et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. Virological.org. Jan. 12, 2021
FUJINO, T. et al. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan. Emerging Infectious Diseases. Feb. 10, 2021.
NAVECA, F. et al. COVID-19 epidemic in the Brazilian state of Amazonas was driven by long-term persistence of endemic SARS-CoV-2 lineages and the recent emergence of the new Variant of Concern P.1. Research Square. Feb. 25, 2021.
RESENDE, P. C. et al. A potential SARS-CoV-2 variant of interest (VOI) harboring mutation E484K in the Spike protein was identified within lineage B.1.1.33 circulating in Brazil. medRxiv. Mar. 13, 2021.
FARIA, N. R. et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv. Mar. 3, 2021.
COUTINHO, R. M. et al. Model-based estimation of transmissibility and reinfection of Sars-CoV-2 P.1 variant. medRxiv. Mar. 9, 2021.
LYTHGOE, K. A. et al. Sars-CoV02 within-host diversity and transmission. Science. Mar. 9, 2021.
MADHI, S. A. et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. New England Journal of Medicine. Mar. 16, 2021.
SOUZA, W. M. et al. Levels of Sars-CoV-2 lineage P.1 neutralization by antibodies elicited after natural infection and vaccination. Preprints with The Lancet. Mar. 1, 2021.
CHEN, R. E. et al. Resistance of Sars-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nature Medicine. Mar. 4, 2021.
LIU, Y. et al. Neutralizing Activity of BNT162b2-Elicited Serum. New England Journal of Medicine. Mar. 8, 2021.
Republish