
riccardo cassiani-ingoni/spl
Astrocyte: producer of the protein that triggers PrPC into actionriccardo cassiani-ingoni/splNothing in nature is free, believed Dr. Ricardo Renzo Brentani. Coupled with keen intuition, that certainty led the oncologist – a pioneer of Brazilian molecular biology until he died in 2011 – to initiate his studies on a protein to which few others gave importance despite its crucial role in brain cell development and defense system balancing.
The protein that Brentani and his collaborators began to investigate 15 years ago goes by the name of cellular prion protein, or PrPC. This protein is produced by the body and can be found on the surface of almost every cell – most abundantly in the immune and central nervous systems. Thanks to the Brazilian researchers, it is now known that the PrPC is a type of gatekeeper to the cells: it organizes and controls the passage of information from the outside to the inside. A noble role indeed for a protein deemed unimportant until only recently. “Nature would not waste the time or the energy to create a protein with no biological function,” said Brentani in 2008, not long after he and his partners in the states of São Paulo, Rio de Janeiro, Minas Gerais and Rio Grande do Sul released the most comprehensive analysis ever published on the functions of the cellular prion protein.
Like a selective magnet anchored to the outside surface of cells, the PrPC attracts specific proteins from the extracellular environment — in some cases, more than one at the same time — and conveys into the cell interior the information they carry. In simple terms, this transfer of information is done in two ways. In one of them, the extracellular protein adheres to the PrPC, which then activates another protein that lies across the cell membrane and triggers chemical signals inside the cell. In the other, PrPC molecules slip into thinner areas of the membrane and are sucked into pouches called vesicles, where they connect to other proteins and relay commands to the nucleus or other regions of the cell. In brain cells, neurons in particular, the chemical signals triggered by the cellular prion protein instruct the cell to stay alive or to issue the elongations that connect it to other neurons (see infographic).
The Brazilian group’s research has provided not only a comprehensive understanding of the activities of the cellular prion protein, but also a new interpretation of the onset and advance of spongiform encephalopathies – as yet incurable diseases caused by structural defects in PrPC. An example of these afflictions is Creutzfeldt-Jakob disease, which takes decades to establish itself in the brain, but evolves and kills within less than a year. As a result of the destruction wreaked by such diseases, the brain turns porous like a sponge.
The knowledge produced in Brazil and the research findings of other countries have also revealed an unexpected connection between these rare and frightening diseases and another, much more prevalent one: Alzheimer’s disease, the fate of one in three people older than 85.

glaucia hajj
Cellular prion protein, marked in green on the neuron surfaceglaucia hajjThe beginning
Brentani saw an opportunity to study the cellular prion protein – and venture into a very competitive field of research – in the early 1990s. At the time, researchers all over the world were investigating the deformed version of PrPC. Known simply as prion, short for proteinaceous infectious particle, the flawed protein was the main suspected cause of a disease detected in parts of England’s beef cattle herd and that gained world notoriety under the name of mad cow disease.
The risk of it being transmissible to humans – the first cases were confirmed in 1996 – launched laboratories into a worldwide race to decipher the infectious protein and what it does. This version of the molecule, which propagates through contact with healthy proteins, causes in humans the type of spongiform encephalopathy described in the 1920s by Hans Gerhard Creutzfeldt and Alfons Maria Jakob. More stable than the cellular prion protein, the deformed molecules clump together, generating long fibers that are toxic to neurons.
While everyone else was studying the defective protein, Brentani decided to investigate the roles of regular, undeformed PrPC. He suspected that a more advanced understanding of how prion diseases establish themselves and evolve – and how they can be fought – would not be possible without knowing how the cellular prion protein works. There was actually some evidence that PrPC was not essential to the body. In 1990 or thereabouts, molecular biologist Charles Weissmann created a lineage of mice that did not produce PrPC, yet appeared perfectly healthy.
But Brentani was not convinced. Years before, he had proposed a theory according to which the same region in the double strand of DNA contained the recipe to produce two proteins – not one. His idea was that the proteins encoded by complementary segments of DNA also played complementary roles and were capable of interacting chemically. But it was a hypothesis that few believed.
Then, in 1991, an American researcher published a letter in Nature saying that, if Brentani were right, the DNA strand complementary to that of the PrPC would contain information about the protein that might possibly trigger it. Brentani, a scholar of proteins associated with cancer, decided to analyze PrPC and its trigger molecule. Alongside biochemist Vilma Martins and biologist Sandro de Souza, then working as researchers at the Ludwig Institute for Cancer Research, in addition to biochemist Vivaldo Moura Neto from the Federal University of Rio de Janeiro (UFRJ), Brentani deduced the structure of that other protein and described it in Nature Medicine in 1997.
 The protein presented by the group – later identified as STI-1, standing for stress inducible protein 1 – was almost twice as large as the cellular prion protein. But nobody knew what either of them did. As they were no experts in neurons, Brentani and Martins invited Rafael Linden, a neuroscientist from UFRJ, to collaborate in the subsequent tests. They discovered that the complex formed by PrPC and STI-1 was fundamental not just for neuronal maturation and elongation, but also to protect neurons from dying (see Pesquisa FAPESP nº 94). Experiments on mice, conducted in partnership with Iván Izquierdo, a researcher at the Pontifical Catholic University of Rio Grande do Sul (PUC/RS), revealed that the cellular prion protein and STI-1 are also crucial for creating memory.
The protein presented by the group – later identified as STI-1, standing for stress inducible protein 1 – was almost twice as large as the cellular prion protein. But nobody knew what either of them did. As they were no experts in neurons, Brentani and Martins invited Rafael Linden, a neuroscientist from UFRJ, to collaborate in the subsequent tests. They discovered that the complex formed by PrPC and STI-1 was fundamental not just for neuronal maturation and elongation, but also to protect neurons from dying (see Pesquisa FAPESP nº 94). Experiments on mice, conducted in partnership with Iván Izquierdo, a researcher at the Pontifical Catholic University of Rio Grande do Sul (PUC/RS), revealed that the cellular prion protein and STI-1 are also crucial for creating memory.
In the immunological system, Linden and his research team showed that the protein complex modulates inflammatory response, either increasing or reducing the activity of defense cells (see Pesquisa FAPESP No. 148). As additional information piled up, such as the role of PrPC in protecting the heart from chemical damage, one thing remained unclear: why the cellular prion protein protected the body’s tissues in certain situations and damaged them in others. An important step was to learn how that protein, which is found on the outer surface of cells, was communicating with the inside.
So Martins, Brentani and Linden appealed to Marco Antonio Prado, then a professor at the Federal University of Minas Gerais, where he was investigating the transport of molecules within cells. In partnership with other researchers, they marked the neuronal cellular prion protein with a fluorescent dye and proceeded to track its movements. They discovered that, once activated by certain proteins, including STI-1, the cellular prion protein glides onto thinner areas of the membrane, temporarily “dips” into the cell and conveys commands to the nucleus or other cellular regions.
The role of STI-1 as a selective magnet or assembly platform for PrPC signaling complexes explained some seemingly controversial experimental results and changed the existing view on how prion diseases establish themselves. Under the new interpretation, in patients with Creutzfeldt-Jakob disease, neurons do not die simply because of adhered prions forming toxic clumps. They also die from the loss of PrPC, which leaves the neurons unprotected from chemical harm.
The findings pertinent to these diseases, the Brazilians suggested, may be applicable to the initial stages of Alzheimer’s. “We started out studying one neurodegenerative disease and discovered connections to others,” says Marco Prado, currently a researcher at the University of Western Ontario, in Canada.
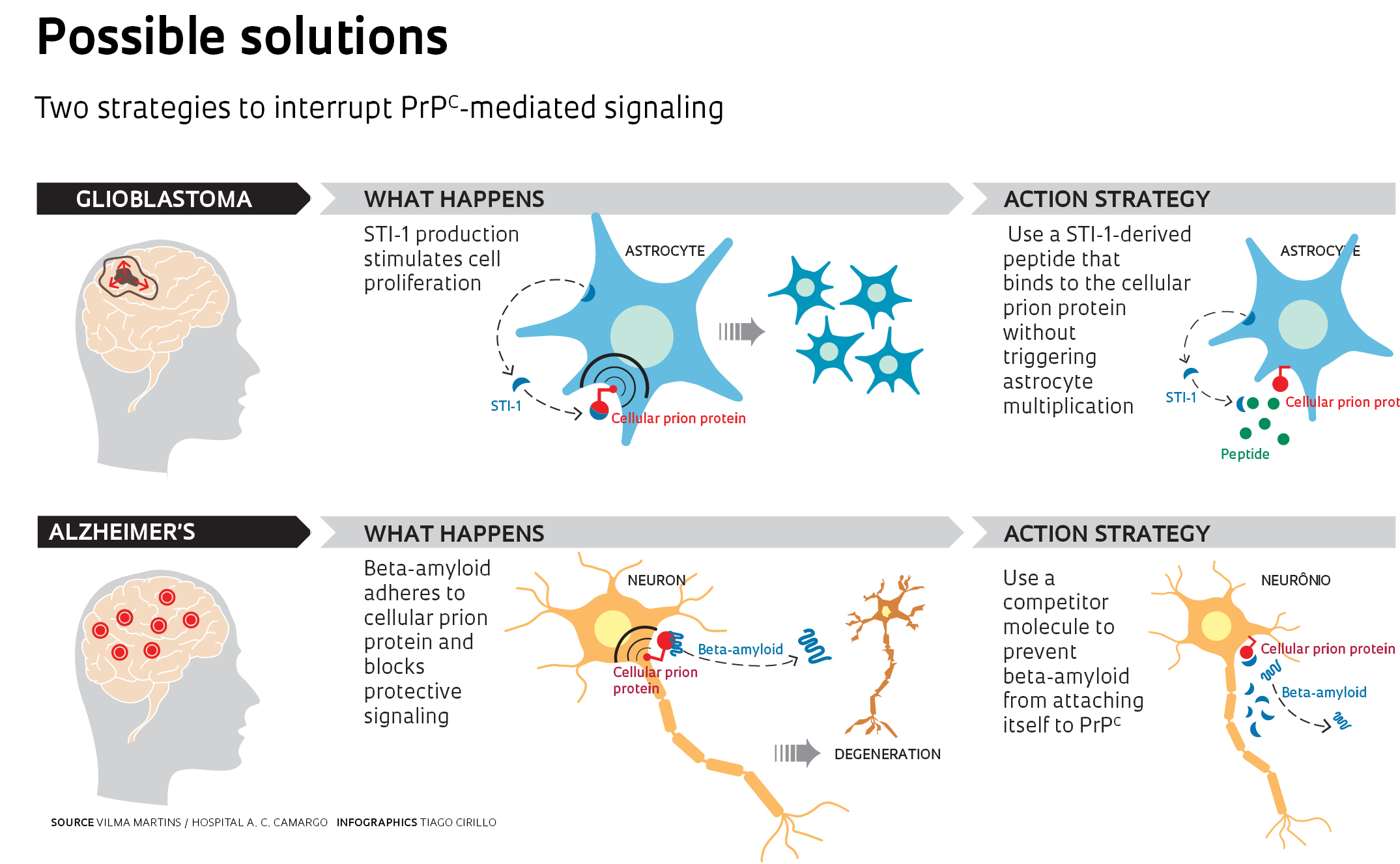
The link between prion diseases and the memory-obliterating illness is that in both cases, PrPC signaling is cut off: in Creutzfeldt-Jakob disease, due to a flaw in the PrPC itself; in Alzheimer’s, due to its activity being blocked by beta-amyloid. “We are not affirming that toxicity does not kill the cell,” explains Vilma Martins, currently a researcher at A. C. Camargo Hospital. “We believe that in addition to this process, the cell also dies because the cellular prion protein is no longer protecting it.”
This view also revealed a new path in the search for strategies to fight these diseases. In a study yet to be published, Martins and Prado tested new ways of interfering with the communications between the PrPC and the beta-amyloid oligomer, a toxic clump of protein fragments that forms in the early stages of Alzheimer’s disease. Through that interference, it might be possible to stop the disease in its tracks. The initial tests have yielded promising results and the researchers have filed a patent application for the use of one of the compounds that prevent oligomer-PrPC interaction (see Pesquisa FAPESP nº 194).
Martins has also joined the fight against glioblastoma, an aggressive type of brain tumor caused by the uncontrolled proliferation of astrocyte-derived cells. Astrocytes are the cells that nourish neurons and defend the central nervous system from invaders, and they’re also responsible for releasing STI-1 into the extracellular environment. In a healthy brain, STI-1 promotes neuronal differentiation and self-renewing in neuronal precursor cells, and also blocks the reproduction of astrocytes. But in glioblastoma, it sets off tumor proliferation.
The strategy chosen by Martins for such cases has been to block PrPC activity with a synthetic fragment of STI-1 that adheres to the cellular prion protein without activating it (see infographic on this page). When tested on mice, the peptide retarded tumoral growth and preserved cognitive ability. For the time being, however, it is not possible to predict whether these strategies will lead to a medication. “What works on animals,” Martins reminds us, “does not always produce the same effects on humans.”
Projects
1. The role of the cellular prion protein in physiological and pathological processes (nº 1999/07124-8) (2000-2004); Grant Mechanism Thematic Project; Coordinator Vilma Regina Martins – Ludwig Institute; Investment R$ 2,353,958.10
2. The role of the cellular prion protein in physiological and pathological processes II (nº 2003/13189-2) (2004-2009); Grant Mechanism Thematic Project; Coordinator Vilma Regina Martins – Ludwig Institute; Investment R$ 1,738,518.72
3. Mechanisms associated with the role of the cellular prion protein and its ligand STI1/Hop: therapeutic approaches (nº 2009/14027-2) (2010-2014); Grant Mechanism Thematic Project; Coordinator Vilma Regina Martins – A. C. Camargo Hospital; Investment R$ 1,699,903.33
Scientific article
LINDEN, R. et al. Physiology of the prion protein. Physiological Reviews. v. 88, p. 673-728, 2008.
From our archives
The neuron link – Issue 94 – December 2003
A fundamental protein – Issue 148 – June 2008
Interruped communication – Issue 194 – April 2012