
desde Río de Janeiro

Léo Ramos
Efecto dañino: moléculas mal enrolladas de la p53 deforman a las proteínas sanas y generan fibras halladas en tumores de piel y de mamaLéo RamosEl término cáncer se aplica a más de una centena de enfermedades ‒algunas que evolucionan rápidamente, otras que sólo se manifiestan luego de décadas; algunas altamente curables, otras irremediablemente fatales‒, todas con una característica en común: la proliferación desenfrenada de las células. Con base en resultados internacionales y datos obtenidos en su laboratorio de la Universidad Federal de Río de Janeiro (UFRJ), el bioquímico Jerson Lima da Silva formuló la hipótesis de que al menos algunos casos de cáncer serían desencadenadas por el mismo mecanismo molecular que provoca la enfermedad de Creutzfeldt-Jakob, la versión en humanos del mal de la vaca loca, que ocasiona la muerte celular precoz y deja al cerebro poroso como una esponja.
En sintonía con ese enfoque, sostenido por Silva y sus colaboradores en un artículo publicado en junio en la revista Bioscience Reports, tanto en el cáncer, signado por la perpetuación de la vida de las células, como en la enfermedad de Creutzfeldt-Jakob, donde la muerte celular se anticipa, el origen del problema sería el mismo: el enrollamiento anormal de una proteína. Puede parecer una causa demasiado sutil para tamaños estragos. Sin embargo, los investigadores consideran que esto tiene sentido. Al fin y al cabo, la estructura tridimensional de esas moléculas grandes y complejas, fundamentales para definir la estructura y el funcionamiento de las células, es la que determina la función que van a desempeñar. Cuando su enrollamiento sale mal, las proteínas generalmente dejan de funcionar como deberían e incluso asumen funciones extras. La diferencia entre los casos de cáncer y los de Creutzfeldt-Jakob radicaría en la proteína afectada.
En las variantes de cáncer analizadas por el grupo de la UFRJ, la deformación afecta a la p53, una proteína a la que se llamó guardiana del genoma porque coordina la reparación del ADN en los casos en que ocurren daños pequeños y porque determina la muerte de la célula cuando esos defectos no pueden repararse. En tanto, en la enfermedad de Creutzfeldt-Jakob, la proteína alterada es el prión celular, una molécula que se adhiere a la superficie externa de las células y controla el tránsito de información desde el medio externo hacia el interior (lea en Pesquisa FAPESP, edición nº 148). En ambos casos, la falla en el enrollamiento parece conferir a la proteína alterada una característica típica de los agentes infecciosos tradicionales, como son los virus y las bacterias: la capacidad de autopropagarse e infectar a otras células.
La idea que apunta que versiones deformadas de una proteína pueden causar enfermedades no es nueva. En la década de 1980 fue postulada por el científico estadounidense Stanley Prusiner para explicar el origen del grupo de enfermedades neurodegenerativas del cual forman parte la enfermedad de Creutzfeldt-Jakob y el mal de la vaca loca, es decir, las encefalopatías espongiformes. Al investigar el agente causante de una encefalopatía que afecta a las ovejas, Prusiner no halló los virus que esperaba. En lugar de ello, tan sólo identificó una proteína defectuosa a la que denominó prion (sigla por partícula proteínica infecciosa) y formuló una explicación al respecto de cómo los priones alterarían a las proteínas sanas. Según esa hipótesis, que le granjeó a Prusiner un premio Nobel en 1997, el simple contacto de la molécula deformada con las proteínas normales resulta suficiente como para inducir una transformación en la estructura tridimensional de las mismas. Se trata de un evento en cadena que, una vez iniciado, no es posible detenerlo, como si fueran fichas de dominó que se tumban. También es un efecto difícil de revertir. Las proteínas defectuosas presentan una estructura más estable que las sanas y se adhieren unas a otras, originando largas fibras tóxicas para las neuronas.
“Creemos que ocurre lo mismo en parte de los casos de cáncer en que la p53 se encuentra alterada”, reveló Silva en su laboratorio a comienzos de agosto, días después de un evento en el que comentó sus resultados con Prusiner. Según Silva, recientes estudios internacionales sugieren incluso que algunas versiones deformadas de la p53 podrían pasar de una célula a otra. Eso no significa, empero, que puedan transmitirse entre individuos de una misma especie. “Esas proteínas serían transmisibles [de una célula a otra], pero no infecciosas”, explicó el bioquímico, quien coordina el Instituto Nacional de Ciencia y Tecnología de Biología Estructural y Bioimagen y es el director científico de la Fundación de Apoyo a la Investigación Científica del Estado de Río de Janeiro (Faperj).
Cáncer de mama
Las evidencias más contundentes acerca de versiones defectuosas de la p53 que pueden actuar como prión ‒los investigadores dicen que ellas manifiestan acción prionoide‒ e inducir la remodelación de las proteínas sanas, llevándolas a perder su función original, surgieron en los últimos dos años. En colaboración con el equipo de la genetista Cláudia Moura-Gallo, de la Universidad del Estado de Río de Janeiro, el grupo de Silva analizó muestras de tumores mamarios de 88 mujeres y constató que en la mayoría de los casos aparecían agregados formados por moléculas anormales de la p53, similares a los agregados amiloides de las enfermedades causadas por prión, ya que esas fibras o agregados también aparecen en otras enfermedades neurodegenerativas, tales como el mal de Parkinson y el de Alzheimer. Las proteínas deformadas generalmente se habían generado a raíz de pequeñas alteraciones en el gen TP53, que contiene la fórmula para la producción de esa proteína.
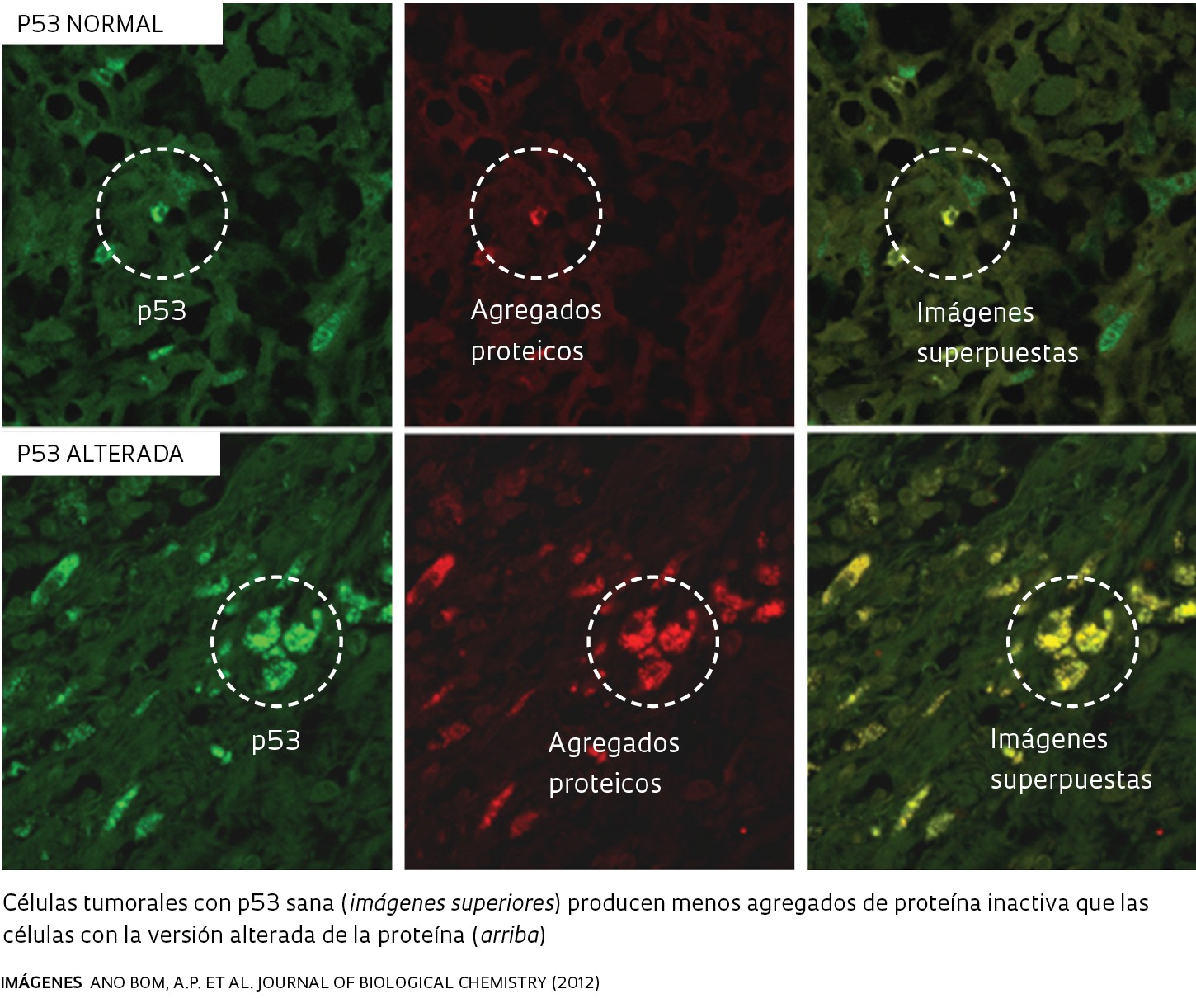
 Infografía Ana Paula Campos Ilustración Pedro HamdanEra la primera vez que se hallaban fibras de p53 en células tumorales. Pero la mera identificación de esas fibras no era suficiente como para demostrar que la proteína alterada podía disparar la deformación de las proteínas sanas, un fenómeno que en inglés recibe el nombre de seeding o siembra, una propiedad peculiar de los priones. En el laboratorio de Silva, la biomédica Ana Paula Ano Bom, la farmacéutica Luciana Rangel y la bioquímica Danielly Costa iniciaron entonces pruebas para identificar las condiciones en que la p53 ‒tanto en su versión sana como en la deformada‒ generaba los agregados. Para ello, midieron el tiempo que tardan en surgir las fibras y la forma que asumen bajo diferentes condiciones químicas (de pH) y físicas (de presión y temperatura).
Infografía Ana Paula Campos Ilustración Pedro HamdanEra la primera vez que se hallaban fibras de p53 en células tumorales. Pero la mera identificación de esas fibras no era suficiente como para demostrar que la proteína alterada podía disparar la deformación de las proteínas sanas, un fenómeno que en inglés recibe el nombre de seeding o siembra, una propiedad peculiar de los priones. En el laboratorio de Silva, la biomédica Ana Paula Ano Bom, la farmacéutica Luciana Rangel y la bioquímica Danielly Costa iniciaron entonces pruebas para identificar las condiciones en que la p53 ‒tanto en su versión sana como en la deformada‒ generaba los agregados. Para ello, midieron el tiempo que tardan en surgir las fibras y la forma que asumen bajo diferentes condiciones químicas (de pH) y físicas (de presión y temperatura).
Y constataron que las dos formas de la p53 generaban espontáneamente los agregados en condiciones similares a las que las células encuentran en el cuerpo humano, con temperatura de 37 grados Celsius y pH neutro o ligeramente ácido; algo típico en los tumores. La formación de esas fibras ocurría incluso cuando en los experimentos se utilizaba tan sólo el segmento central, la porción de la P53 que interactúa con el ADN. La diferencia radica en que ambos agregados tóxicos aparecieron más rápidamente a partir de la p53 alterada, según informaron los investigadores en agosto de 2012 en el Journal of Biological Chemistry (vea la infografía en la parte superior).
El resultado más importante surgió cuando las investigadoras agregaron diminutas concentraciones de la proteína defectuosa en recipientes que contenían p53 sana. A semejanza de un remolino, la proteína deformada atrajo a las moléculas normales e indujo su conversión en la forma alterada, confirmando aquella propiedad de seeding. La capacidad para inducir la deformación resultó mayor cuando se utilizó la versión de la p53 alterada como resultado de una mutación denominada R248Q, una de las siete que aparecen con mayor frecuencia en los casos de cáncer. “Hallamos agregados de esa forma mutante en muestras de cáncer de mama y en linajes de células de este tumor cultivadas en laboratorio”, comenta Danielly.
Pequeñas alteraciones, grandes efectos
La formación de los aglomerados de p53 parece no ser exclusiva de los tumores mamarios. Al comienzo de este año, el equipo de Rakez Kayed, de la Universidad de Texas, en Galveston, describió esas fibras en un tipo bastante frecuente de tumor de piel, o carcinoma basocelular. Recientemente, el grupo de Silva también las identificó en muestras de glioblastoma humano, uno de los tumores cerebrales más agresivos que se conocen.
 La tendencia a la agregación no sólo ocurre como consecuencia de la mutación R248Q. Otras alteraciones puntuales en el gen crean versiones de la proteína propensas a agregarse, informaron los investigadores de la UFRJ. Más allá de modificar la estructura de la p53 sana, las versiones deformadas de esa molécula captan y dañan a otras dos proteínas de la misma familia: la p63, que controla la multiplicación celular, y la p73, que, en forma independiente, orienta a las células con defecto hacia la apoptosis, un tipo de muerte programada. “Esas dos proteínas también desempeñan un rol relevante como antitumoral”, explica Danielly.
La tendencia a la agregación no sólo ocurre como consecuencia de la mutación R248Q. Otras alteraciones puntuales en el gen crean versiones de la proteína propensas a agregarse, informaron los investigadores de la UFRJ. Más allá de modificar la estructura de la p53 sana, las versiones deformadas de esa molécula captan y dañan a otras dos proteínas de la misma familia: la p63, que controla la multiplicación celular, y la p73, que, en forma independiente, orienta a las células con defecto hacia la apoptosis, un tipo de muerte programada. “Esas dos proteínas también desempeñan un rol relevante como antitumoral”, explica Danielly.
En un estudio publicado en julio en la PLoS One, Xavier Roucou y su equipo en la Universidad de Sherbrooke, en Quebec, Canadá, demostraron que una versión defectuosa de la p53 es, en efecto, capaz de infectar células sanas. Los investigadores agregaron moléculas deformadas en cultivos de células y observaron que las proteínas alteradas eran absorbidas por bolsas que se formaban en la membrana. Ya en el interior de las células, la p53 mal ovillada desencadenó la formación de fibras.
Como toda idea nueva, la hipótesis que sostiene que las versiones mutantes de la p53 pueden funcionar como prión no tiene consenso. “El trabajo del equipo de la UFRJ es bastante consistente, pero se necesitan más evidencias”, comenta la bioquímica Vilma Martins, del Centro Internacional de Investigaciones del A. C. Camargo Cáncer Center, en São Paulo, estudiosa de las enfermedades causadas por priones. Una de las etapas que faltan para demostrar la acción prionoide de la p53 consiste en evaluar si la proteína modificada en laboratorio provoca tumores en modelos animales. A pesar de la cautela, Martins considera que ese mecanismo podría explicar el origen de una parte de los casos de cáncer relacionados con las mutaciones en el gen TP53.
Si la idea del grupo de la UFRJ fuera correcta, contribuirá a la comprensión del desarrollo de los tumores considerados espontáneos, que no se transmiten de una generación a otra y surgen como consecuencia de alteraciones genéticas en el embrión ya formado o en el individuo adulto. La acción prionoide de la p53 sería una buena explicación para los tumores espontáneos ‒la gran mayoría de los casos de cáncer‒, especialmente cuando aparece la denominada dominancia negativa. “En ese fenómeno biológico, la sola alteración de una de las dos copias de un gen resulta suficiente como para conducir al desarrollo de una enfermedad”, explica Silva, quien ya en 2003 analizó esa posibilidad para el caso de la p53, cuando comenzó a estudiar el enrollamiento de dicha proteína. E imagina la posibilidad de utilizar las fibras de p53 en un test, como marcador molecular de la gravedad del cáncer o como pronóstico. E incluso, que en el futuro sea posible interferir en ese mecanismo e intentar detener el desarrollo de algunos tumores. “Quizá”, dice, “pueda hallarse una forma de bloquear el proceso de agregación.
Artículos científicos
SILVA, J. L. et al. Expanding the prion concept to cancer biology: dominant-negative effect of aggregates of mutant p53 tumor suppressor. Bioscience Reports. 27 jun. 2013
SILVA, J. L. et al. Ligand binding and hydration in protein misfolding: insights from studies of prion and p53 tumor suppressor proteins. Accounts of Chemical Research. v. 43, n.2, p. 271-9. 2010.
ANO BOM, A. P. et al. Mutant p53 aggregates into prion-like, amyloid oligomers and fibrils: implications for cancer. Journal of Biological Chemistry. v. 287, n. 33, p. 28.152-62. 10 ago. 2012.