Desde Monte Santo, Bahía

Eduardo CesarLa vía sacra de Monte Santo: un sitio de peregrinación que recibe miles de personas cada añoEduardo Cesar
José de Andrade Pereira es un hombre de carácter. En 2004, llevó a su hijo mayor, que a los tres años era de baja estatura, tenía dedos cortos, cabeza grande y dificultades en el habla ‒y otra vez sufría un fuerte dolor de oído‒, a un centro de salud de Monte Santo, en el interior de Bahía. El médico le dijo que, amén de tratarlo por su dolor de oído, no podría hacer nada más ante una enfermedad que no conocía y que su pronóstico era solamente aguardar que muriera. Pereira reaccionó: “¡Eso es algo que nunca voy a hacer!”. Realizó el viaje de seis horas hasta Salvador y le preguntó a un portero del Hospital Universitario Profesor Edgard Santos a quién podía consultarle por el tratamiento para un niño como el suyo. Los médicos examinaron al niño y luego a su hermano de 11 meses, en el viaje siguiente, y determinaron que ambos padecían mucopolisacaridosis tipo VI, una rara enfermedad de origen genético para la que, por entonces, no había tratamiento. Pereira advirtió: “Hay otros niños así por allá”. Y su visión del mundo cambió la historia de esa ciudad del sertón bahiano.
Monte Santo fue un campamento de las tropas del gobierno que lucharon en la Guerra de Canudos. En la plaza principal se exhibe una escultura en madera de Antonio Conselheiro, el beato que lideró a sus seguidores campesinos, tenidos como opositores a la república brasileña naciente. Engalanando la escultura hay una matadeira, el cañón utilizado en las batallas donde murieron 25 mil sediciosos y 5 mil soldados. En los últimos años, Monte Santo se ha convertido en escenario de otras batallas: la detección, el tratamiento y la prevención de enfermedades genéticas raras, que comenzaron a conocerse a partir de la advertencia de Pereira. Antes de eso, los niños con enfermedades tales como la mucopolisacaridosis se quedaban en sus casas. Sus padres consideraban que no había nada que hacer.

Eduardo CesarLos exvotos de la capilla al final del sendero de piedraEduardo Cesar
Médicos y científicos de Salvador, Río de Janeiro y Porto Alegre visitaron Monte Santo por primera vez en 2006 y se asombraron con la diversidad de enfermedades raras que detectaban en un solo lugar. Han diagnosticado a 13 personas con mucopolisacaridosis tipo VI, una proporción 240 veces mayor que la del promedio nacional, 84 con deficiencia auditiva con posible origen genético, 12 con hipotiroidismo congénito, nueve con fenilcetonuria, una afección que puede causar deficiencia intelectual si no se la trata, cuatro con osteogénesis imperfecta, signada por la extrema fragilidad de los huesos, y cuatro con síndrome de Treacher Collins, que afecta el desarrollo de los huesos del cráneo.
Se cree que los casamientos entre parientes, antaño muy frecuentes, podrían haber propiciado el surgimiento de enfermedades físicas y mentales de origen genético. Muchas personas se casaban sin saber que tenían ascendientes cercanos en común. José Pereira y su esposa, Júlia Isaura dos Santos Pereira, supieron que eran parientes recién hace pocos años, al reconstruir el árbol genealógico de sus familias junto a los investigadores de Salvador, y finalmente comprendieron por qué habían oído comentarios sobre tíos que padecían la misma enfermedad detectada en sus dos hijos mayores. Tal vez las raíces más profundas de estos problemas se encuentren en la propia historia del lugar. Varios relatos del historiador bahiano José Calasans señalan que el municipio, hoy en día con 52 mil habitantes ‒diseminados por 47 poblados alrededor del núcleo urbano‒ fue un centro de convergencia de individuos enfermos que acudían en busca de los milagros de Conselheiro, lo que apuntaló la fama religiosa del lugar. Él fue quien reformó las capillas a lo largo de la vía sacra, un camino agreste y sinuoso de piedras, con 2,8 kilómetros (km) de extensión, que conduce a una capilla que se alza en la cima de un cerro, construida en 1786 por un cura italiano. Cada año, miles de peregrinos suben por el camino de piedras, a veces de rodillas o con una piedra sobre la cabeza, para cumplir promesas. Por su habitual condición de pobreza, los enfermos, curados o no, sus familiares y romeros, pueden haber afrontado dificultades para regresar a sus tierras de origen, o bien, prefirieron quedarse en la zona.

Eduardo CesarMonte Santo al anochecer: escenario de batallas históricasEduardo Cesar
Como se estima que habría más gente aún no diagnosticada, la médica genetista Angelina Acosta, docente de la Universidad Federal de Bahía (UFBA), se presentó ante el Concejo Deliberante de Monte Santo al comienzo de la tarde del día 10 de julio y expuso, ante médicos y políticos, su plan para realizar un censo de salud en toda la población. “Pertenecemos a las universidades, pero trabajamos junto a ustedes, para que nuestro trabajo tenga una aplicación práctica”, afirmó. La secretaria municipal de Salud, Itácia Macedo de Andrade Silva, escuchaba atentamente. “Quiero hacer algo por mi tierra”, dijo ella, explicando por qué regresó a la ciudad luego de estudiar enfermería en Salvador. A la mañana siguiente, el equipo coordinado por Acosta y Kiyoko Sandes conversó con 80 agentes comunitarios de salud que visitarán los poblados a partir de este mes, en busca de otros casos. El diálogo trajo a la luz que había gente que aún no había sido examinada y se creó un comité de apoyo para la realización del censo y el tratamiento. José Pereira era uno de sus integrantes.
Las enfermedades raras configuran un mundo de sufrimiento, soledad, fantasías y culpa, que recién ahora comienza a analizarse públicamente. “El Sistema Único de Salud [SUS] reconoció que las enfermedades raras deben tratarse”, comenta Clarice Alegre Petramale, directora del departamento de gestión e incorporación de tecnologías del Ministerio de Salud. La política nacional de atención médica a las personas con enfermedades raras ‒a las que se define como cualquier enfermedad que padezcan al menos 13 individuos en cada grupo de 20 mil‒ se encuentra en vigencia desde mayo de este año. Los grupos de enfermedades con atención prioritaria se anunciarán para fin de año.
 Infografía: Ana PaulaPara la mayoría de las enfermedades raras no existen medicamentos específicos, tan sólo hay un tratamiento de sostén, que incluye fisioterapia y fonoaudiología. Cuando existe alguna medicación, ésta generalmente es importada y se obtiene por medio de decisiones judiciales. “Los remedios son caros y muchas veces con eficacia incierta, pues no han pasado por todas los ensayos rigurosos de evaluación, puesto que se los probó en un número muy reducido de pacientes”, dice Magda Carneiro-Sampaio, directora del Instituto del Niño de la Universidad de São Paulo (USP). “Y además, los medicamentos suelen prescribirse cuando las enfermedades cursan fases avanzadas, y entonces ya no son tan eficaces. Es algo que está mal barajado, pero eso no ocurre solamente en Brasil”. Maira Catharina Ramos, de la Universidad de Brasilia, calculó que si el gobierno comprara voluntariamente tan sólo uno de los fármacos utilizados para el tratamiento de la mucopolisacaridosis, ello implicaría un ahorro de 50 millones de reales, en comparación con lo que se gasta para cumplir con los mandatos judiciales, que obligan al gobierno a adquirir los remedios. Se estima que en el país habría 13 millones de personas con alguna enfermedad rara, de las cuales, en el mundo, ya se han descrito 6 mil tipos, en su gran mayoría, de origen genético.
Infografía: Ana PaulaPara la mayoría de las enfermedades raras no existen medicamentos específicos, tan sólo hay un tratamiento de sostén, que incluye fisioterapia y fonoaudiología. Cuando existe alguna medicación, ésta generalmente es importada y se obtiene por medio de decisiones judiciales. “Los remedios son caros y muchas veces con eficacia incierta, pues no han pasado por todas los ensayos rigurosos de evaluación, puesto que se los probó en un número muy reducido de pacientes”, dice Magda Carneiro-Sampaio, directora del Instituto del Niño de la Universidad de São Paulo (USP). “Y además, los medicamentos suelen prescribirse cuando las enfermedades cursan fases avanzadas, y entonces ya no son tan eficaces. Es algo que está mal barajado, pero eso no ocurre solamente en Brasil”. Maira Catharina Ramos, de la Universidad de Brasilia, calculó que si el gobierno comprara voluntariamente tan sólo uno de los fármacos utilizados para el tratamiento de la mucopolisacaridosis, ello implicaría un ahorro de 50 millones de reales, en comparación con lo que se gasta para cumplir con los mandatos judiciales, que obligan al gobierno a adquirir los remedios. Se estima que en el país habría 13 millones de personas con alguna enfermedad rara, de las cuales, en el mundo, ya se han descrito 6 mil tipos, en su gran mayoría, de origen genético.
De norte a sur
El trabajo de los investigadores académicos junto a los profesionales de la salud locales en busca de otros habitantes con enfermedades infrecuentes sitúa a Monte Santo a la cabeza del Censo Nacional de Aislados (Ceniso), organizado por el Instituto Nacional de Genética Médica y Poblacional (Inagemp). En abril de 2014, un artículo publicado en la revista Genetics and Molecular Biology presentó uno de los resultados del Ceniso: un estudio nacional de los municipios con grupos de individuos o familias con enfermedades genéticas. Una versión actualizada de este mapeo, que se muestra en la página anterior, engloba a 81 localidades, donde residen 4.136 personas con características genéticas específicas, los denominados aislados genéticos. No siempre se trata de enfermedades. El municipio sureño de Cândido Godói, por ejemplo, reúne una cifra extraordinaria de gemelos. El equipo de la médica genetista Lavínia Schuler Faccini, docente de la Universidad Federal de Rio Grande do Sul (UFRGS) y miembro del comité coordinador del Inagemp, identificó 91 casos de mellizos y uno de trillizos nacidos entre 1959 y 2000 en ese municipio, que hoy cuenta con 6 mil habitantes. Entre 1994 y 2006, el 2% de las niñas nacidas en Cândido Godói correspondió a gemelas, el doble del promedio nacional. En uno de los distritos, la tasa de gemelos llegó al 10%.
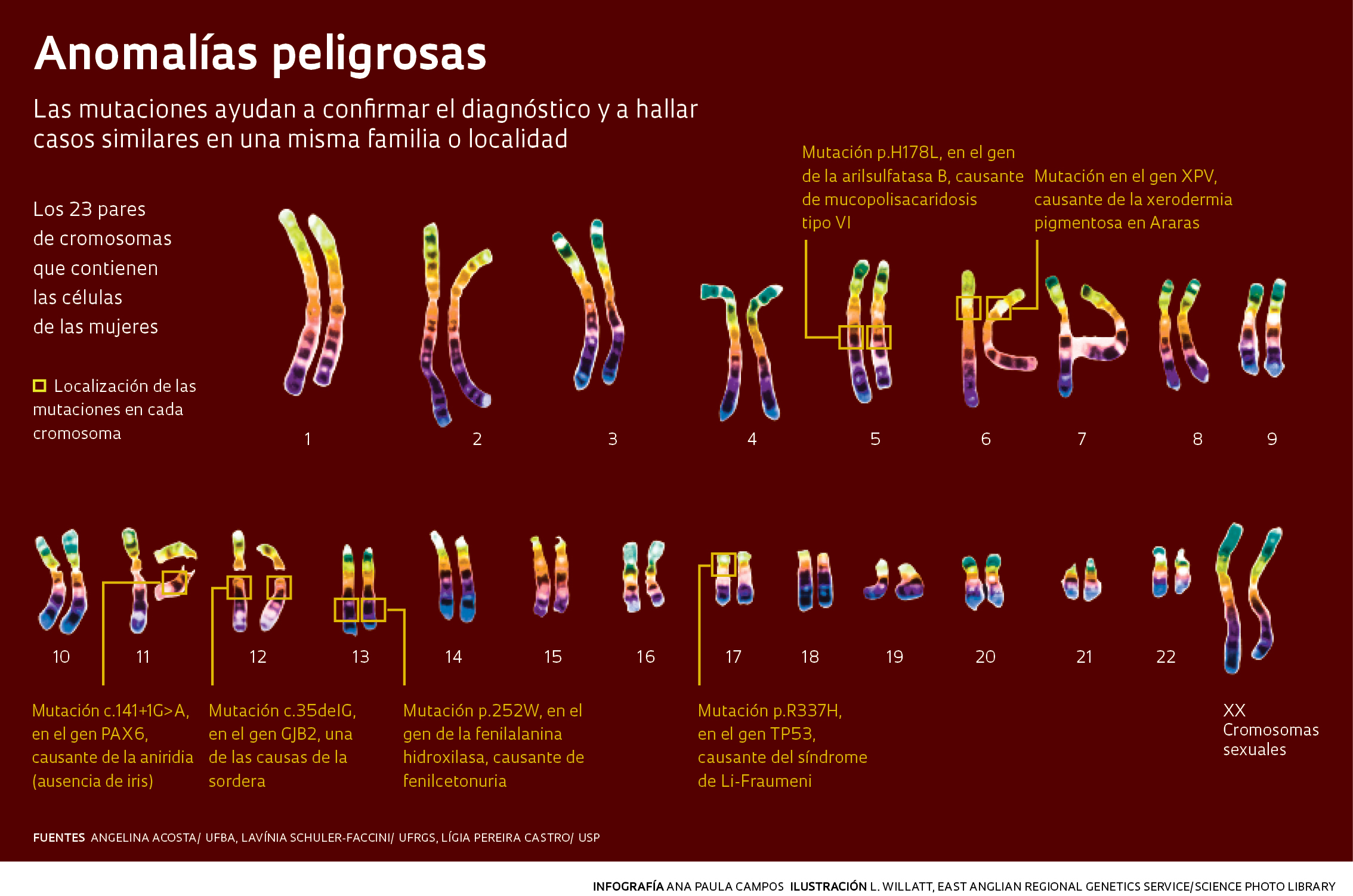 Infografía: Ana Paula Campos / Ilustración: L. WILLATT, EAST ANGLIAN REGIONAL GENETICS SERVICE/ SCIENCE PHOTO LIBRARYAlgunas enfermedades genéticas se manifiestan en la adultez, incluso después de los 40 años de edad, como sucede en el caso de la ataxia de Machado-Joseph, que provoca una progresiva pérdida del equilibrio, de los movimientos y del habla. Faccini y su equipo, quienes realizan asesoría genética para los familiares de casi 400 pacientes con ataxia en Porto Alegre, han notado que el diagnóstico, generalmente tardío, suele generar angustia y culpa, pues para entonces la gente pudo haber tenido hijos o nietos con las mutaciones causantes de la enfermedad.
Infografía: Ana Paula Campos / Ilustración: L. WILLATT, EAST ANGLIAN REGIONAL GENETICS SERVICE/ SCIENCE PHOTO LIBRARYAlgunas enfermedades genéticas se manifiestan en la adultez, incluso después de los 40 años de edad, como sucede en el caso de la ataxia de Machado-Joseph, que provoca una progresiva pérdida del equilibrio, de los movimientos y del habla. Faccini y su equipo, quienes realizan asesoría genética para los familiares de casi 400 pacientes con ataxia en Porto Alegre, han notado que el diagnóstico, generalmente tardío, suele generar angustia y culpa, pues para entonces la gente pudo haber tenido hijos o nietos con las mutaciones causantes de la enfermedad.
Existen enfermedades cuyo alcance es regional, como por ejemplo el síndrome de Li-Fraumeni, un trastorno genético hereditario que predispone al cáncer, identificado en 325 personas de los estados de Río de Janeiro, São Paulo, Paraná, Santa Catarina y Rio Grande do Sul. Un estudio reciente registró una elevada incidencia, del 0,27%, de la mutación causante de la enfermedad en 171 mil bebés analizados en la ciudad de Curitiba, lo cual indica que esa dolencia, en ciertos lugares, no es rara y sería necesario un seguimiento intensivo, fundamentalmente en los individuos con mayor riesgo.

Eduardo CesarPrimas con fenilcetonuria controlada: al lado, Raíra Anielli Carvalho Silva, entre su madre, Eliana Batista Carvalho, y su hermano, Ranieri Carvalho Silva (izquierda), el el abuelo de 92 años, José Lopes de Carvalho, y el padre, José Nildo Andrade SilvaEduardo Cesar
El médico Eduardo Enrique Castilla, investigador de la Fiocruz de Río de Janeiro y uno de los impulsores del censo, estima que el total de municipios donde ya se han detectados enfermedades raras en Brasil representaría tan sólo el 20% de lo previsto. La lista no cesa de crecer, porque los investigadores siguen hallando indicios de otros sitios que aún no han sido mapeados. En el mes de junio de este año, el genetista Carlos Menck, de la USP, viajó a Miracatu, una ciudad con 30 mil habitantes ubicada a 130 km de São Paulo, e identificó a cuatro personas de una misma familia con xerodermia pigmentosa, que ya fue diagnosticada en 22 habitantes de un pueblo en el interior del estado de Goiás. Si bien la enfermedad es la misma, las mutaciones que la causan, los genes y los cromosomas afectados son distintos entre la gente de los dos estados.
A partir de 2013, el equipo de la genetista Denise Cavalcanti, de la Universidad de Campinas (Unicamp), identificó nuevos clusters, otra denominación para los aislados genéticos, de diferentes displasias óseas, enfermedades que afectan el crecimiento de los huesos. Los clusters se encuentran en cinco localidades de los estados de Ceará, Alagoas, Pernambuco y São Paulo. Uno de ellos, identificado con la colaboración de una médica genetista de Fortaleza, impresiona por la cantidad de individuos diagnosticados hasta ahora, que suma 27, en 22 familias. En al menos 10 pequeñas ciudades del interior de Ceará se distribuye en forma dispersa una enfermedad bastante rara denominada picnodisostosis, la misma que padeció el artista francés Henri de Toulouse-Lautrec, determinante de su baja estatura. El grupo de Cavalcanti trabaja ahora en la identificación del posible lugar de origen de la mutación que causa esa dolencia, el denominado efecto fundador, en Ceará.

Eduardo CesarCamilly Vitória de Souza Andrade, entre el padre, José Armando Moraes de Andrade, y la madre, Cremilda Maria de Souza AndradeEduardo Cesar
“No se imagina lo importante que es para las madres saber el nombre de la enfermedad que padecen sus hijos, aunque no exista tratamiento, porque es entonces cuando dejan de deambular de un médico a otro”, dice Cavalcanti. Cierto día, ella recibió una carta remitida por una mujer de Belém que le agradeció por el diagnóstico de un hijo y comentaba: “Era angustiante no contar con un diagnóstico, era como andar sin rumbo o a tientas en la oscuridad”. Cuando se enteran del nombre de las enfermedades, las madres “vuelven a tener expectativas por sus hijos”, afirma Isabella Queiroz, docente de la Escuela Bahiana de Medicina y psicóloga de la Apae de Salvador, que atiende a las familias con enfermedades genéticas de Monte Santo. “Hemos realizado más de 200 sesiones de asesoría genética”. En esas entrevistas, el equipo médico les explica que los trastornos genéticos hereditarios ocurren por la transmisión de genes con alteraciones (o mutaciones), porque su origen no es únicamente el casamiento entre parientes. Al analizar un mapa genético de la familia relacionando los individuos sanos y enfermos, una mujer comprendió a su manera lo que estaba ocurriendo: “¿Se sumaron un defecto mío con otro de mi marido y nació fallado, es así?”. También es ahí cuando descargan su culpa por haber tenido hijos enfermos, su desamparo y su enojo. Uno de los hombres le preguntó al profesional que lo atendía: “¿No puedo tener más hijos? ¿Entonces estoy condenado?”. El equipo de asesoramiento genético sabe que debe informarles los riesgos que presentan las enfermedades hereditarias sin intervenir en la decisión de las parejas con respecto a tener o no tener hijos.
A veces, las parejas jóvenes, antes de tener hijos, consultan voluntariamente al servicio médico para detectar eventuales mutaciones perjudiciales, lo cual indica que el número de casos de algunas enfermedades podría reducirse en los próximos años. El hábito de casarse con primos ‒mucho más frecuente en los países musulmanes que en Brasil‒ posiblemente sea lo más difícil de modificar, porque ha sido una costumbre adoptada hace mucho tiempo, ya sea como forma de mantener las tierras en posesión de la familia o bien por preferencias personales. Cuando visitaron Tabuleiro do Norte, un municipio situado a 200 km de Fortaleza con alta incidencia de una enfermedad metabólica conocida como síndrome de Gaucher, los investigadores oyeron que los hombres decían que las mujeres de la capital eran buenas como novias y las de Tabuleiro, para casarse. Por lo tanto, es posible que circunscribirse al propio lugar sea la causa del elevado índice de enfermedades genéticas. Se comenta que un cura de Monte Santo ofrecía bicicletas para que los muchachos buscaran novias en otros sitios, pero nadie aceptó.

Eduardo CesarLibro de casamientos de la parroquia de Monte Santo en 1938: en caso que los contrayentes fueran primos, el vicario debía otorgarles la “dispensa de impedimento”Eduardo Cesar
“Han cambiado muchas cosas”, señala Maria Olívia Sousa Costa, secretaria de Salud de Monte Santo entre 2008 y 2011. “Los médicos asumieron la responsabilidad de identificar las enfermedades raras, y las madres adquirieron conciencia de sus derechos a la salud y reclamaron cuando faltaba la infusión”. La infusión es un preparado inyectable que contiene la enzima arilsulfatasa B, que los niños con mucopolisacaridosis tipo VI no producen y se les aplica en sesiones semanales de cuatro horas de duración. Durante años, los padres llevaban a los niños a Salvador para recibir la medicación, que a partir de 2011 se les aplica en un anexo del hospital de la ciudad. Pero no todo ha cambiado, pues no siempre se dispone de la medicación. “Siempre hay que estar reclamando, pero nunca desisto, eso no”, dice Pereira. Sus dos hijos comenzaron el tratamiento en 2008, en Salvador, cuatro años después del diagnóstico, puesto que aún no se había aprobado el uso de la enzima en Brasil. La enfermedad es irreversible. Actualmente, Jorge tiene 17 años y su hermano Sidney, 14, ambos con estaturas menores a un metro y con dificultades motrices. Vitor Gonzaga Andrade, el hijo de Alaíde Gonzaga Andrade, prima de Pereira, comenzó a recibir la medicación a los dos años y ahora, ya con seis, corre como un niño sano.
Cremilda Maria de Souza Andrade quedó consternada al enterarse que su hija Camilly tenía fenilcetonuria, diagnosticada al mes de su nacimiento. Viajó a Salvador e inició el tratamiento, que consiste en una dieta rigurosa, sin ninguna proteína que contenga fenilalanina, sustancia que el organismo no logra procesar. “Ahora estoy feliz”, dice Souza Andrade, mostrando con orgullo los cuadernos de su hija, que hoy tiene 11 años y cumple permanentemente con su dieta. Camilly es prima de Raíra Anielli Carvalho Silva, de 12 años, quien también padece fenilcetonuria, afección que le detectaron ni bien nació y que se la controlaron por medio de la alimentación. Sin embargo, en el mismo pueblo donde ella reside, viven dos hermanos, hoy con 40 y 27 años de edad respectivamente, que portan la misma mutación que las niñas. Como la prueba del talón no era común en la región en la época en que ellos nacieron, les detectaron la enfermedad mucho más tardíamente, y al no habérsela controlado, les causó los desórdenes mentales que ahora les impiden salir de su casa.
Trabajo de campo
Para realizar un trabajo inclusivo, los investigadores deben salir del laboratorio, ponerse ropa apropiada, viajar por lugares inimaginables, conocer los hábitos y los silencios de los residentes en el interior del país y buscar información en otros lugares. Para localizar a los individuos con mayor riesgo de padecer enfermedades genéticas, Angelina Acosta y su equipo consultaron las actas de matrimonio, bautismos y defunciones en la parroquia de Monte Santo desde 1831, y elaboraron el historial de 1.419 familias. Luego de perderse, hallaron el camino, valioso incluso para los historiadores, al percatarse que las mujeres obtenían un apellido tan sólo después de casarse, las parejas anotaban a las hijas con el apellido de la madre y al hijo con el del padre, además se utilizaban apodos como nombres, como en el caso de un hombre conocido como Santana, que en realidad se llamaba José da Silva. En Monte Santo, los nombres y apellidos se repetían formando un laberinto similar al de la familia Buendía en Cien años de soledad, cuyos integrantes vivían casándose entre sí y luego temían que les nacieran hijos con cola de cerdo.
La publicación de artículos científicos sigue siendo algo importante, pero no es prioritaria, porque hay “una obligación moral” de informarles los hallazgos en primer lugar a las comunidades estudiadas, subraya Castilla. Durante la mañana de un sábado, Faccini y su equipo se pusieron al frente de 200 personas en el salón de fiestas de la parroquia de Cândido Godói para explicarles el motivo del elevado número de gemelos, muchos de ellos entre el público: probablemente era la consecuencia de una alteración del gen de la proteína p53, que podía propiciar el desarrollo de dos embriones en la gestación. Durante décadas, se creyó que los gemelos eran consecuencia de un efecto producido por el agua ‒supuestamente especial‒ del municipio.
Artículos científicos
CASTILLA, E. E.; SCHULER-FACCINI, Lavínia. From rumors to genetic isolates. Genetics and Molecular Biology. v. 1, n. 37, p. 186-93. 2014.
TAGLIANI-RIBEIRO A. et al. High twinning rate in Cândido Godói: a new role for p53 in human fertility. Human Reproduction. v. 27, n. 9, p. 2866-71. 2012.
MACHADO, T. M. B. et al. Types of marriages, population structure and genetic disease. Journal of Biosocial Science. v. 45, n. 4, p. 461-70. 2013.