Al cabo de un año de la pandemia decretada oficialmente por la Organización Mundial de la Salud (OMS) el 11 de marzo de 2020, la humanidad asistió atónita al contagio de 122 millones de personas y a la muerte de al menos 2,7 millones, la tercera parte de estas últimas en tan solo tres países: Estados Unidos, Brasil y México. Asimismo, pudo seguirse prácticamente en tiempo real y con un nivel de precisión tal vez nunca visto, la evolución del patógeno que puso de rodillas al sistema sanitario y afectó profundamente a la economía de muchas naciones. Desde su identificación a finales de 2019 en la ciudad de Wuhan, en la región central de China, el virus Sars-CoV-2 ha sufrido una serie de transformaciones mientras se propagaba por el planeta. A medida que infectaba progresivamente cada vez a más personas y se replicaba, fue acumulando pequeñas modificaciones en su material genético hasta que, en distintos lugares y momentos, ya era tan diferente al original que pasó a considerárselo una nueva variante que, al prosperar y diseminarse, empezó a denominársela como un linaje. Un linaje agrupa a ejemplares con un mismo origen y muy similares entre sí, pero que pueden tener pequeñas diferencias.
El 15 de marzo de este año, Nextstrain, una herramienta de visualización de genomas virales, enumeró nada menos que 359 linajes del nuevo coronavirus catalogados desde diciembre de 2019 por el sistema de clasificación Pangolin (lea en la página 24). Entre tantos, tres de ellos –uno que surgió en el Reino Unido, otro en Sudáfrica y un tercero en Brasil– han generado especial preocupación porque son más transmisibles, pueden eludir la acción de los anticuerpos y, en algunos casos, causan una enfermedad más grave que los que circulaban antes. “Con las herramientas genéticas disponibles en la actualidad, estamos observando la evolución de este patógeno a medida que esto ocurre”, dice el virólogo José Luiz Proença Módena, coordinador del Laboratorio de Estudio de Virus Emergentes de la Universidad de Campinas (Unicamp).
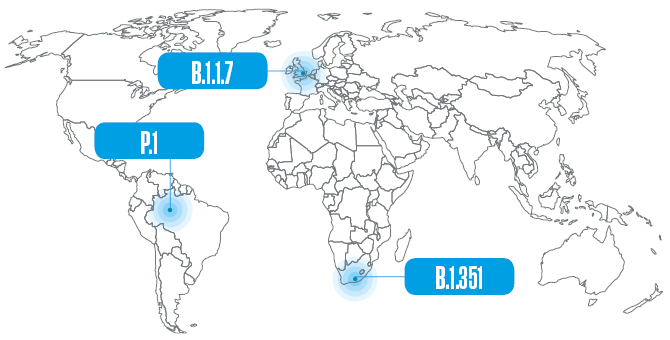
La primera de estas variantes, que ya se ha convertido en un linaje y que en marzo estaba presente en 118 países, se detectó el 14 de diciembre del año pasado en el Reino Unido. Comenzó a circular en el mes de septiembre en el condado de Kent, en el sudeste de Inglaterra, y se propagó rápidamente. Inicialmente apodada como la variante Kent o británica, más tarde se la empezó a identificar con un frío y sobrio conjunto de letras y números (B.1.1.7) definido por una nomenclatura propuesta por investigadores de Australia y del Reino Unido. Esta secuencia alfanumérica indica que es la séptima variante derivada de la primera que desciende del linaje B.1, uno de los dos que surgieron originalmente en Wuhan; el otro, posiblemente más antiguo, es el A.1, que desapareció a mediados del año pasado. A causa de algunas alteraciones (mutaciones) que presenta en su genoma, la variante B.1.1.7 se transmite al menos dos veces más fácilmente que el linaje que la originó y, según parece, también ocasiona una enfermedad más grave. A finales de 2020 ya había indicios de que podría contribuir a un aumento de las internaciones hospitalarias en el Reino Unido, y ahora se han detectado evidencias de su asociación con un mayor riesgo de muerte.
En un estudio publicado el día 15 de marzo en la revista Nature, el epidemiólogo Nicholas Davies, junto a matemáticos y estadísticos de la Escuela de Higiene y Medicina Tropical de Londres, estimaron que una persona infectada con esta variante tiene, en promedio, un 61 % más de probabilidades de morir que alguien que haya contraído cualquiera de los linajes que circulaban anteriormente. Los científicos arribaron a esta conclusión tras analizar 4.945 óbitos ocurridos en un grupo de 1.146.534 británicos que habían dado positivo en el test para el nuevo coronavirus. Días antes, un grupo de investigadores dirigidos por el médico Robert Challen, de la Universidad de Exeter, también en el Reino Unido, presentaron un resultado similar en un artículo que salió publicado el 10 de marzo en la revista The BMJ. Al comparar el total de fallecidos de un grupo de 54.906 británicos infectados con la variante B.1.1.7 con las muertes registradas para igual cantidad de personas que habían sido infectadas por otro linaje del virus, comprobaron que el primer grupo se enfrentaba a un riesgo de muerte un 64 % mayor que el segundo.
Pese a que se propaga fácilmente entre la población humana, en la fase inicial de la pandemia el Sars-CoV-2 parecía ser un virus relativamente manejable, que mutaba muy lentamente. En promedio, cada mes acumulaba dos mutaciones en la secuencia de casi 30 mil bases nitrogenadas, las letras químicas que componen su genoma, una especie de manual de instrucciones para la fabricación de nuevas copias del virus. Para tener un parámetro comparativo, el influenzavirus A, causante de las pandemias de gripe, evoluciona mucho más rápido: sufre una mutación cada vez que se multiplica, en cuestión de horas, un ritmo que obliga a actualizar anualmente la composición de la vacuna contra la gripe.
Hacia el final de 2020, cuando se anunciaron los resultados de la eficacia de las primeras vacunas y renacía la esperanza de que la pandemia amainara, el nuevo coronavirus volvió a sorprender. “Empezaron a aparecer variantes que presentaban simultáneamente a varias mutaciones y se diseminaron rápidamente, sustituyendo a las anteriores”, recuerda el virólogo Fernando Spilki, investigador de la Universidad Feevale, en Rio Grande do Sul, y coordinador de la Red Nacional de Ómicas del Covid-19, la llamada Corona-ómica BR, que monitorea la circulación del virus en el país.
Una de estas variantes es la que originó el linaje B.1.1.7. La misma presenta 23 mutaciones con respecto a la B.1 de Wuhan, de las cuales 17 provocan un cambio en un aminoácido en las proteínas del virus; las otras seis son inocuas (vea la infografía debajo). Los aminoácidos son compuestos químicos que, unidos en una secuencia, conforman las proteínas. En algunos casos, el reemplazo de un solo aminoácido es suficiente para alterar la estructura tridimensional de la proteína y modificar su funcionamiento.
El linaje B.1.351 se detectó inicialmente en el área metropolitana de Nelson Mandela Bay, en el sur de Sudáfrica, en octubre de 2020, aunque posiblemente haya surgido meses antes y fue notificado por las autoridades del país recién en diciembre. Descrito por el grupo coordinado por el virólogo brasileño Túlio de Oliveira, de la Universidad KwaZulu-Natal, en un artículo publicado en marzo de este año en la revista Nature, fue detectado con mayor frecuencia en individuos jóvenes y sanos, y ahora ya circula localmente en Europa y en América del Norte.
Los virus de este linaje también presentan 23 modificaciones en su genoma, ocho de ellas en el gen de la proteína S. Además de la N501Y, otras dos mutaciones en la spike alertan a los expertos: la E484K, que representa el reemplazo de un glutamato (E) por una lisina (K) y que en febrero de este año también comenzó a detectarse en el linaje británico, y la K417N, en la que se sustituye una lisina por una asparagina (N). Estas sustituciones alteran una región de la spike denominada dominio de unión al receptor (RBD, por sus siglas en inglés), que entra en contacto más estrechamente con el receptor ACE2 en la superficie de las células humanas y allana el camino para la entrada del virus, e impiden que ciertos anticuerpos producidos tras la aplicación de algunas vacunas o por las infecciones previas con otras variantes del Sars-CoV-2 se conecten al virus y lo neutralicen. Los estudios preliminares indican que las personas infectadas con B.1.351 –la segunda variante preocupante detectada en diciembre de 2020, según la PHE– acumulan una mayor cantidad de virus en su organismo, lo que podría facilitar la transmisión, así como beneficiarse potencialmente menos de los efectos protectores de algunas vacunas.
Estas tres alteraciones de la spike viral también están presentes en el linaje P.1 originario de Brasil, que también presenta otras siete alteraciones en la misma proteína. Esta variante surgió a partir de la B.1.1.28, circulante en el país desde el comienzo de la pandemia, y fue nombrada con otra letra porque el sistema solo acepta tres conjuntos de guarismos. El linaje brasileño posiblemente comenzó a circular en Manaos a principios de noviembre y fue detectado al mes siguiente, en forma simultánea, por investigadores de la USP y de la Fundación Oswaldo Cruz (Fiocruz), cuando sobrevino el aumento del número de hospitalizaciones que marcó el inicio de la segunda ola de la pandemia en el norte del país, el sistema de salud en el estado de Amazonas colapsó y hubo gente que murió por asfixia como consecuencia de la falta de oxígeno medicinal en los hospitales.
Un individuo infectado por el P.1 produce, en promedio, el doble de virus que quienes se contaminaron con los linajes que circulaban previamente en Brasil, según pudo constatar el equipo del virólogo Renato Santana de Aguiar, de la Universidad Federal de Minas Gerais (UFMG), miembro de la red Corona-ómica BR. La presencia de una mayor cantidad de virus en el organismo aumenta de 1,4 a 2,2 veces la posibilidad de transmisión, según el cálculo estimado por el grupo encabezado por Sabino y el biomédico portugués Nuno Faria, de la Universidad de Oxford, en el Reino Unido. Otro equipo de la USP, coordinado por el biólogo Paulo Inácio Prado, también comprobó que el P.1 tiene una probabilidad –baja, es cierto, de un 6,4 %– de volver a infectar a quienes ya se habían contagiado con otros linajes del virus.
Algunos expertos sostienen que, dadas estas características del P.1, se deberían haber adoptado medidas más drásticas de salud pública a principios de año para tratar de evitar la propagación de este linaje, tales como un estricto control de la circulación de personas y el bloqueo de los vuelos desde el estado de Amazonas a otras regiones. Sin embargo, desde el mes de diciembre, al menos 120.000 personas abandonaron Amazonas con destino a otros estados y países, e incluso hubo pacientes con covid-19 que fueron trasladados a otras regiones brasileñas por falta de camas.

Christopher Furlong/Getty Images
Ciudadanos de Manchester, en el Reino Unido, realizan testeos en febrero de este año, luego del brote con el linaje B.1.1.7
Christopher Furlong/Getty ImagesUna de las consecuencias que esto generó es que, para mediados de marzo, el linaje P.1 ya representaba el 41 % de las infecciones provocadas por el nuevo coronavirus en Brasil (véase el gráfico de la página 18). Otro análisis que llevó a cabo la Red Genómica de la Fiocruz indicó que en menos de tres meses esta variante fue la responsable de más de la mitad de las infecciones en seis estados: Pernambuco (el 51 %), Rio Grande do Sul (el 63 %), Río de Janeiro (el 63 %), Santa Catarina (el 64 %), Paraná (el 70 %) y Ceará (el 71 %). “Todo el país está viviendo ahora una situación similar a la que atravesó Manaos en el mes de diciembre, con el P.1 en plena propagación”, dice Sabino, quien coordina a nivel local el Centro Conjunto Brasil-Reino Unido para la Detección, el Diagnóstico, la Genómica y la Epidemiología de los Arbovirus (Cadde), financiado por la agencia británica Medical Research Council y la FAPESP.
“Brasil se ha transformado en un terreno fértil para la aparición de variantes y linajes más contagiosos”, dice el virólogo Eurico Arruda, de la USP de Ribeirão Preto, quien lleva casi tres décadas estudiando los coronavirus (lea en Pesquisa FAPESP, edición nº 301). La población susceptible de contraer el virus es numerosa, la vacunación en el país es muy lenta y las medidas de prevención, como el distanciamiento social y el uso de mascarillas, no se han adoptado con eficacia en todas las regiones. “Cuanta más gente infecte el virus, más se multiplicará y más sufrirá alteraciones”, explica.
Los virus son microorganismos naturalmente propensos a acumular mutaciones en su genoma, pues, a diferencia de los seres vivos formados por células, no disponen de mecanismos eficaces para corregir los errores. A medida que el material genético viral se replica para generar nuevas copias del virus, se producen alteraciones aleatorias, de una manera similar a las que ocurren cuando alguien copia a mano un texto muy extenso (aunque lo ejecute con suma atención). Sin proteínas que detecten eficazmente las imprecisiones y las desbaraten, las mutaciones acaban incorporándose al material genético de los nuevos virus que, en el caso del Sars-CoV-2, es una molécula de ARN de cadena simple.
Rodger Bosch/AFP vía Getty Images
Profesional de la salud efectúa controles a un paciente con covid-19 en Sudáfrica, tras la aparición de una nueva variante
Rodger Bosch/AFP vía Getty Images“Las mutaciones constituyen algo habitual, pero no todas son importantes para incrementar la transmisibilidad o la patogenicidad del virus”, explica la bioquímica Marilda Siqueira, del Laboratorio de Virus Respiratorios y Sarampión, de la Fiocruz, un instituto referente en vigilancia genómica del coronavirus para el Ministerio de Salud y la OMS. Otras, sin embargo, pueden conferirle al virus una ventaja adaptativa y le permiten, por ejemplo, penetrar con mayor facilidad en las células, multiplicarse más o eludir la acción de los anticuerpos. En estos casos, quedan incorporadas en el genoma del virus y se transmiten a las generaciones futuras.
“Sospechamos que este es el caso de las mutaciones N501Y, E484K y K417N, que aparecieron de manera independiente en las variantes que surgieron en diferentes lugares del mundo y parecen acrecentar la aptitud viral”, dice Aguiar, de la UFMG. Además de estar presentes en el linaje P.1, en el B.1.351 de origen sudafricano y desde principios de año también en el B.1.1.7 británico, aparece igualmente en el material genético de otras dos variantes brasileñas, identificadas con la ayuda del grupo de Siqueira: la P.2, que ya se diseminó por todo el país, y la N.9, detectada a principios de marzo. Todavía se las está investigando y, por ahora, no son tan preocupantes como el linaje P.1.
“Pienso que, con el tiempo, surgirán variantes más transmisibles, pero que causen una enfermedad más leve, como ocurrió con el virus de la gripe A, que causó la pandemia de gripe española en 1918”, dice Arruda. Un análisis de la diversidad genética del Sars-CoV-2 en 1.313 personas, publicado el 9 de marzo en la revista Science, elaborado por la epidemióloga Katrina Lythgoe y colaboradores, de la Universidad de Oxford, sugiere que el desarrollo de variantes más transmisibles que escapan a la acción del sistema de defensa es inusual. Sin embargo, una vez que emergen, pueden propagarse rápidamente.
Además de permitirle al virus propagarse con mayor facilidad, las mutaciones detectadas en los nuevos linajes han generado preocupación en muchos expertos por la amenaza que pueden suponer para la eficacia de las vacunas. Los inmunizantes desarrollados por las empresas farmacéuticas estadounidenses Janssen y Novavax, por ejemplo, ya han mostrado una menor eficacia contra la variante sudafricana B.1.351. Al igual que Novavax, la vacuna desarrollada en conjunto por la Universidad de Oxford y la empresa farmacéutica anglosueca AstraZeneca, que también pierde parte de su efecto contra el linaje B.1.1.7, procedente del Reino Unido. No obstante, su rendimiento es mucho peor contra el linaje sudafricano. Se observó una importante reducción en su capacidad de prevenir los casos leves y moderados de la enfermedad causada por el B.1.351 (no hubo casos graves en el grupo vacunado ni en el que recibió placebo), según muestran los resultados de un ensayo clínico con alrededor de 2.000 participantes que salió publicado el 16 de marzo en la revista New England Journal of Medicine.
Michael Dantas/AFP vía Getty Images
Un paciente derivado para su hospitalización en Manaos, en Brasil, durante el mes de enero, en medio de una nueva ola de contagios
Michael Dantas/AFP vía Getty ImagesLos estudios realizados por el grupo de Módena en la Unicamp, y por el equipo del inmunólogo Michael Diamond, de la Universidad de Washington en Saint Louis, Estados Unidos, identificaron, respectivamente, cierto nivel de pérdida de efecto de los anticuerpos producidos por la CoronaVac y por la vacuna de Pfizer contra la variante P.1 en pruebas in vitro. Empero, aún no puede afirmarse que estas variantes esquiven el efecto de las vacunas en las personas. La literatura científica sobre el tema aún muestra contradicciones y otros estudios han llegado a conclusiones distintas. De todas maneras, a finales del mes de febrero algunos fabricantes de vacunas anunciaron que están trabajando en dosis de refuerzo y en otras fórmulas que contemplan las mutaciones de las nuevas variantes.
Asimismo, los anticuerpos no solamente funcionan acoplándose al virus y bloqueando su entrada en las células. También pueden desencadenar reacciones químicas perjudiciales para las partículas virales o actuar como marcadores, permitiendo su reconocimiento por las células de defensa que los fagocitan y los digieren. “La evaluación de otras estrategias del sistema inmunitario, tal como es el caso de la actividad de las células de defensa, es más laboriosa, pero es esencial para arribar a conclusiones más sólidas”, comenta el biomédico William Souza, quien realiza una pasantía posdoctoral en la Universidad de Oxford y colaboró en los experimentos de Módena y Sabino. “Las vacunas siguen evitando que la gente enferme y muera”.
Para Módena, los resultados obtenidos hasta ahora contra los nuevos linajes del virus refuerzan la idea de que incluso aquellos que ya han padecido la enfermedad o han sido vacunados deben mantener los cuidados, seguir utilizando mascarillas y adoptando medidas de higiene y distanciamiento social. “De momento, lo que se sabe es que quienes ya han tenido covid-19 o incluso los que han recibido algunas de las vacunas pueden reinfectarse con el virus de uno de los linajes preocupantes”, dice. En una encuesta realizada en enero por la revista Nature a la que respondieron cien virólogos, inmunólogos y expertos en enfermedades infecciosas de diferentes países, el 90 % dijo que suponían que el virus podría tornarse endémico y seguir circulando en pequeños nichos durante los próximos años, ya sea porque escapa a la acción de los anticuerpos, porque no habrá vacunas suficientes para todos, porque algunos se niegan a vacunarse o porque siguieron infectando a animales en la naturaleza.
El surgimiento de los linajes La secuenciación del genoma permite realizar un seguimiento de las transformaciones del virusPara monitorear el surgimiento de variantes y linajes, los expertos secuencian el material genético de los virus circulantes en cierto momento y en una región determinada y lo comparan con el que existía anteriormente. Hasta comienzos de marzo, se habían secuenciado en todo el mundo casi 670 mil genomas del Sars-CoV-2; y unas 4.000 de esas secuenciaciones se hicieron en Brasil. En la actualidad, el país cuenta con al menos tres redes de laboratorios que realizan monitoreo genómico en forma sistemática: la Red Genómica de la Fiocruz, financiada por el Ministerio de Salud nacional, la Red Nacional de Ómicas de Covid-19 (Corona-ómica BR), con el respaldo del Ministerio de Ciencia, Tecnología e Innovación, y el Centro Conjunto Brasil-Reino Unido para la Detección, el Diagnóstico, la Genómica y la Epidemiología de los Arbovirus (Cadde), que se mantiene con fondos aportados por la agencia británica Medical Research Council y por la FAPESP. “Necesitamos ampliar nuestra capacidad de secuenciación y estamos encarando ese proyecto”, comenta la bioquímica Marilda Siqueira, investigadora de la Red Genómica de la Fiocruz. “Así y todo, hemos sido capaces de detectar la irrupción del linaje británico en varios estados y registramos el inicio de la circulación del P.1, el P.2 y el N.9”. La secuenciación del genoma demanda el uso de reactivos importados, demora días de trabajo y cuesta alrededor de 600 reales. Para mejorar la vigilancia, algunos laboratorios brasileños han diseñado test moleculares (PCR) que permiten detectar los nuevos linajes del virus. Uno de ellos es el de la Fiocruz en el estado de Amazonas. Otro es el del virólogo Renato Santana Aguiar, de la UFMG, que identifica las variantes P.1, P.2, B.1.1.7 y B.1.351. “El test tiene un costo de 60 reales y está listo en algunas horas”, relata Santana Aguiar. “Nos permitirá conocer con mayor precisión la circulación de esos linajes”. Sin embargo, la determinación del genotipo mediante el PCR no reemplaza a la secuenciación, que es la única forma de identificar el surgimiento de nuevas variantes y linajes.
Dándole un nombre al virus Los sistemas adoptan distintas estrategias para denominar a los linajesQuienes hayan leído algo sobre los nuevos linajes y variantes del nuevo coronavirus se habrán topado con secuencias de letras y números que parecen no tener sentido. Son intentos de organizar el conocimiento disponible sobre el virus y seguir de cerca su difusión y su evolución. En los últimos meses se conocieron al menos tres estrategias para nombrarlos. Una de ellas es un sistema propuesto por el biólogo Andrew Rambaut, de la Universidad de Edimburgo, en Escocia, al cual se lo conoce por la sigla Pangolin, acrónimo de Phylogenetic Assignment of Named Global Outbreak Lineages. Este sistema provee información detallada sobre la circulación del virus en un momento determinado. En el mismo, las variantes o linajes se clasifican según el grado de parentesco evolutivo, atribuyéndoles una letra seguida por hasta tres números: por ejemplo, el linaje que se originó en el Reino Unido es el B.1.1.7, la séptima variante derivada de la primera que deriva del linaje B.1, uno de los dos que surgieron originalmente en Wuhan. El linaje sudafricano es el B.1.351 (el 351º descendiente del B.1). En tanto, el brasileño, por ser descendiente del linaje B.1.1.28, que ya tenía tres números, recibió una nueva letra y se convirtió en el P.1. Un segundo sistema de clasificación es el Nextstrain, propuesto por el grupo de Trevor Bedford y sus colaboradores, del Centro de Investigación del Cáncer Fred Hutchinson, de Estados Unidos. Este sistema agrupa a los virus en conjuntos más grandes (clados), que son descritos por el año de identificación del clado, seguido de una letra y el nombre de una mutación que define al grupo. Así, el linaje del Reino Unido forma parte del clado 20I/501Y.V1, el de Sudáfrica es 20H/501.V2 y el de Brasil, 20J/501.V3. El tercer sistema de clasificación proviene de un consorcio internacional creado en 2008 cuyo nombre es Gisaid. Este ha sido el que ha adoptado la OMS, y también agrupa a los virus por clados, definidos por una o más letras que integran las mutaciones principales del grupo. Allí el linaje del Reino Unido se incluye en el clado GRY, el de Sudáfrica en el GH y el de Brasil, en el GR.
Proyectos
1. Centro conjunto Brasil-Reino Unido para el descubrimiento, el diagnóstico, la genómica y la epidemiología de arbovirus (Cadde) (nº 18/14389-0); Modalidad Proyecto Temático; Investigadora responsable Ester Cerdeira Sabino (USP); Inversión R$ 5.331.725,16.
2. ICTP – Instituto sudamericano de física fundamental. Un centro regional de física teórica (nº 16/01343-7); Modalidad Proyecto Temático; Investigador responsable Nathan Jacob Berkovits (Unesp); Inversión R$ 15.421.379,38.
3. Patogénesis y neurovirulencia de virus emergentes en Brasil (nº 16/00194-8); Modalidad Joven Investigador; Investigador responsable José Luiz Proença Módena (Unicamp); Inversión R$ 2.509.395,80.
4. Caracterización de factores de riesgo intrínsecos y desarrollo de nuevas alternativas de diagnóstico y tratamiento del covid-19 (nº 20/04558-0); Modalidad Ayuda de Investigación – Regular; Investigador responsable José Luiz Proença Módena (Unicamp); Inversión R$ 184.077,00.
5. Replicación y efectos celulares de rinovirus en tejidos linfoides (nº 18/25605-6); Modalidad Ayuda de Investigación – Regular; Investigador responsable Eurico de Arruda Neto (FMRP-USP); Inversión R$ 236.000,00.
Artículos científicos
DAVIES, M. G. et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. 15 mar. 2021.
CHALLEN, R. et al. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. The BMJ. 10 mar. 2021.
TEGALLY, H. et al. Emergence of a SARS-CoV-2 variant of concern with mutations in spike glycoprotein. Nature. 9 mar. 2021.
NAVECA, F. et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein. Virological.org. 11 ene. 2021.
FARIA, N. R. et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. Virological.org. 12 ene. 2021
FUJINO, T. et al. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan. Emerging Infectious Diseases. 10 feb. 2021.
NAVECA, F. et al. COVID-19 epidemic in the Brazilian state of Amazonas was driven by long-term persistence of endemic SARS-CoV-2 lineages and the recent emergence of the new Variant of Concern P.1. Research Square. 25 feb. 2021.
RESENDE, P. C. et al. A potential SARS-CoV-2 variant of interest (VOI) harboring mutation E484K in the Spike protein was identified within lineage B.1.1.33 circulating in Brazil. medRxiv. 13 mar. 2021.
FARIA, N. R. et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv. 3 mar. 2021.
COUTINHO, R. M. et al. Model-based estimation of transmissibility and reinfection of Sars-CoV-2 P.1 variant. medRxiv. 9 mar. 2021.
LYTHGOE, K. A. et al. Sars-CoV02 within-host diversity and transmission. Science. 9 mar. 2021.
MADHI, S. A. et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. New England Journal of Medicine. 16 mar. 2021.
SOUZA, W. M. et al. Levels of Sars-CoV-2 lineage P.1 neutralization by antibodies elicited after natural infection and vaccination. Pre-prints with The Lancet. 1º mar. 2021.
CHEN, R. E. et al. Resistance of Sars-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nature Medicine. 4 mar. 2021.
LIU, Y. et al. Neutralizing Activity of BNT162b2-Elicited Serum. New England Journal of Medicine. 8 mar. 2021.
Republicar